ZITHROMAX® IV
(azithromycin dihydrate IV)
Find ZITHROMAX® IV medical information:
Find ZITHROMAX® IV medical information:
ZITHROMAX® IV Quick Finder
17 PATIENT COUNSELING INFORMATION
17 PATIENT COUNSELING INFORMATION
Patients should be informed of the following serious and potentially serious adverse reactions that have been associated with ZITHROMAX
Diarrhea: Inform patients that diarrhea is a common problem caused by antibacterial drugs which usually ends when the antibacterial is discontinued. Sometimes after starting treatment with antibacterials, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibacterial. If this occurs, patients should notify their physician as soon as possible.
Highlights
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ZITHROMAX safely and effectively. See full prescribing information for ZITHROMAX. ZITHROMAX (azithromycin) for injection, for intravenous use Initial U.S. Approval: 1991 RECENT MAJOR CHANGES
INDICATIONS AND USAGEZITHROMAX is a macrolide antibacterial drug indicated for mild to moderate infections caused by designated, susceptible bacteria: To reduce the development of drug-resistant bacteria and maintain the effectiveness of ZITHROMAX and other antibacterial drugs, ZITHROMAX should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. (1.3) DOSAGE AND ADMINISTRATIONDOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions are nausea (4%), diarrhea (4%), abdominal pain (3%), or vomiting (1%). (6) To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSUSE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 1/2022 |
Indications and Usage
1 INDICATIONS AND USAGE
ZITHROMAX (azithromycin) for injection is a macrolide antibacterial drug indicated for the treatment of patients with infections caused by susceptible strains of the designated microorganisms in the conditions listed below.
1.1 Community-Acquired Pneumonia
due to Chlamydophila pneumoniae, Haemophilus influenzae, Legionella pneumophila, Moraxella catarrhalis, Mycoplasma pneumoniae, Staphylococcus aureus, or Streptococcus pneumoniae in patients who require initial intravenous therapy.
1.2 Pelvic Inflammatory Disease
due to Chlamydia trachomatis, Neisseria gonorrhoeae, or Mycoplasma hominis in patients who require initial intravenous therapy. If anaerobic microorganisms are suspected of contributing to the infection, an antimicrobial agent with anaerobic activity should be administered in combination with ZITHROMAX.
ZITHROMAX for injection should be followed by ZITHROMAX by the oral route as required. [see Dosage and Administration (2)]
1.3 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of ZITHROMAX (azithromycin) and other antibacterial drugs, ZITHROMAX (azithromycin) should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
Dosage and Administration
2 DOSAGE AND ADMINISTRATION
[see Indications and Usage (1) and Clinical Pharmacology (12.3)]
2.1 Community-Acquired Pneumonia
The recommended dose of ZITHROMAX for injection for the treatment of adult patients with community-acquired pneumonia due to the indicated organisms is 500 mg as a single daily dose by the intravenous route for at least two days. Intravenous therapy should be followed by azithromycin by the oral route at a single, daily dose of 500 mg, administered as two 250 mg tablets to complete a 7- to 10-day course of therapy. The timing of the switch to oral therapy should be done at the discretion of the physician and in accordance with clinical response.
2.2 Pelvic Inflammatory Disease
The recommended dose of ZITHROMAX for injection for the treatment of adult patients with pelvic inflammatory disease due to the indicated organisms is 500 mg as a single daily dose by the intravenous route for one or two days. Intravenous therapy should be followed by azithromycin by the oral route at a single, daily dose of 250 mg to complete a 7-day course of therapy. The timing of the switch to oral therapy should be done at the discretion of the physician and in accordance with clinical response.
2.3 Preparation of the Solution for Intravenous Administration
The infusate concentration and rate of infusion for ZITHROMAX for injection should be either 1 mg/mL over 3 hr or 2 mg/mL over 1 hr. ZITHROMAX for injection should not be given as a bolus or as an intramuscular injection.
Reconstitution
Prepare the initial solution of ZITHROMAX for injection by adding 4.8 mL of Sterile Water for Injection to the 500 mg vial, and shaking the vial until all of the drug is dissolved. Since ZITHROMAX for injection is supplied under vacuum, it is recommended that a standard 5 mL (non-automated) syringe be used to ensure that the exact amount of 4.8 mL of Sterile Water is dispensed. Each mL of reconstituted solution contains 100 mg azithromycin. Reconstituted solution is stable for 24 hr when stored below 30°C (86°F).
Parenteral drug products should be inspected visually for particulate matter prior to administration. If particulate matter is evident in reconstituted fluids, the drug solution should be discarded.
Dilute this solution further prior to administration as instructed below.
Dilution
To provide azithromycin over a concentration range of 1–2 mg/mL, transfer 5 mL of the 100 mg/mL azithromycin solution into the appropriate amount of any of the diluents listed below:
Normal Saline (0.9% sodium chloride)
1/2 Normal Saline (0.45% sodium chloride)
5% Dextrose in Water
Lactated Ringer's Solution
5% Dextrose in 1/2 Normal Saline (0.45% sodium chloride) with 20 mEq KCl
5% Dextrose in Lactated Ringer's Solution
5% Dextrose in 1/3 Normal Saline (0.3% sodium chloride)
5% Dextrose in 1/2 Normal Saline (0.45% sodium chloride)
Normosol-M in 5% Dextrose
Normosol-R in 5% Dextrose
| Final Infusion Solution Concentration (mg/mL) | Amount of Diluent (mL) |
|---|---|
| 1 mg/mL | 500 mL |
| 2 mg/mL | 250 mL |
Other intravenous substances, additives, or medications should not be added to ZITHROMAX for injection, or infused simultaneously through the same intravenous line.
Dosage Forms and Strengths
Contraindications
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
Serious allergic reactions, including angioedema, anaphylaxis, and dermatologic reactions including Acute Generalized Exanthematous Pustulosis (AGEP), Stevens-Johnson Syndrome, and toxic epidermal necrolysis have been reported in patients on azithromycin therapy. [see Contraindications (4.1)]
Fatalities have been reported. Cases of Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) have also been reported. Despite initially successful symptomatic treatment of the allergic symptoms, when symptomatic therapy was discontinued, the allergic symptoms recurred soon thereafter in some patients without further azithromycin exposure. These patients required prolonged periods of observation and symptomatic treatment. The relationship of these episodes to the long tissue half-life of azithromycin and subsequent prolonged exposure to antigen is unknown at present.
If an allergic reaction occurs, the drug should be discontinued and appropriate therapy should be instituted. Physicians should be aware that the allergic symptoms may reappear after symptomatic therapy has been discontinued.
5.2 Hepatotoxicity
Abnormal liver function, hepatitis, cholestatic jaundice, hepatic necrosis, and hepatic failure have been reported, some of which have resulted in death. Discontinue azithromycin immediately if signs and symptoms of hepatitis occur.
5.3 Infantile Hypertrophic Pyloric Stenosis (IHPS)
Following the use of azithromycin in neonates (treatment up to 42 days of life), IHPS has been reported. Direct parents and caregivers to contact their physician if vomiting or irritability with feeding occurs.
5.4 QT Prolongation
Prolonged cardiac repolarization and QT interval, imparting a risk of developing cardiac arrhythmia and torsades de pointes, have been seen with treatment with macrolides, including azithromycin. Cases of torsades de pointes have been spontaneously reported during postmarketing surveillance in patients receiving azithromycin. Providers should consider the risk of QT prolongation, which can be fatal when weighing the risks and benefits of azithromycin for at-risk groups including:
- patients with known prolongation of the QT interval, a history of torsades de pointes, congenital long QT syndrome, bradyarrhythmias or uncompensated heart failure
- patients on drugs known to prolong the QT interval
- patients with ongoing proarrhythmic conditions such as uncorrected hypokalemia or hypomagnesemia, clinically significant bradycardia, and in patients receiving Class IA (quinidine, procainamide) or Class III (dofetilide, amiodarone, sotalol) antiarrhythmic agents.
Elderly patients may be more susceptible to drug-associated effects on the QT interval.
5.5 Cardiovascular Death
Some observational studies have shown an approximately two-fold increased short-term potential risk of acute cardiovascular death in adults exposed to azithromycin relative to other antibacterial drugs, including amoxicillin. The five-day cardiovascular mortality observed in these studies ranged from 20 to 400 per million azithromycin treatment courses. This potential risk was noted to be greater during the first five days of azithromycin use and does not appear to be limited to those patients with preexisting cardiovascular diseases. The data in these observational studies are insufficient to establish or exclude a causal relationship between acute cardiovascular death and azithromycin use. Consider balancing this potential risk with treatment benefits when prescribing ZITHROMAX.
5.6 Clostridioides difficile-Associated Diarrhea
Clostridioides difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including ZITHROMAX (azithromycin for injection), and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antibacterial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibacterial use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibacterial treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
5.7 Exacerbation of Myasthenia Gravis
Exacerbations of symptoms of myasthenia gravis and new onset of myasthenic syndrome have been reported in patients receiving azithromycin therapy.
5.8 Infusion Site Reactions
ZITHROMAX for injection should be reconstituted and diluted as directed and administered as an intravenous infusion over not less than 60 minutes. [see Dosage and Administration (2)]
Local IV site reactions have been reported with the intravenous administration of azithromycin. The incidence and severity of these reactions were the same when 500 mg azithromycin was given over 1 hour (2 mg/mL as 250 mL infusion) or over 3 hr (1 mg/mL as 500 mL infusion) [see Adverse Reactions (6)]. All volunteers who received infusate concentrations above 2 mg/mL experienced local IV site reactions and, therefore, higher concentrations should be avoided.
Adverse Reactions
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Hypersensitivity [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Infantile Hypertrophic Pyloric Stenosis (IHPS) [see Warnings and Precautions (5.3)]
- QT Prolongation [see Warnings and Precautions (5.4)]
- Cardiovascular Death [see Warnings and Precautions (5.5)]
- Clostridioides difficile-Associated Diarrhea (CDAD) [see Warnings and Precautions (5.6)]
- Exacerbation of Myasthenia Gravis [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In clinical trials of intravenous azithromycin for community-acquired pneumonia, in which 2 to 5 IV doses were given, the reported adverse reactions were mild to moderate in severity and were reversible upon discontinuation of the drug. The majority of patients in these trials had one or more co-morbid diseases and were receiving concomitant medications. Approximately 1.2% of the patients discontinued intravenous ZITHROMAX therapy, and a total of 2.4% discontinued azithromycin therapy by either the intravenous or oral route because of clinical or laboratory side effects.
In clinical trials conducted in patients with pelvic inflammatory disease, in which 1 to 2 IV doses were given, 2% of women who received monotherapy with azithromycin and 4% who received azithromycin plus metronidazole discontinued therapy due to clinical side effects.
Clinical adverse reactions leading to discontinuations from these studies were gastrointestinal (abdominal pain, nausea, vomiting, diarrhea), and rashes; laboratory side effects leading to discontinuation were increases in transaminase levels and/or alkaline phosphatase levels.
Overall, the most common adverse reactions associated with treatment in adult patients who received IV/Oral ZITHROMAX in studies of community-acquired pneumonia were related to the gastrointestinal system with diarrhea/loose stools (4.3%), nausea (3.9%), abdominal pain (2.7%), and vomiting (1.4%) being the most frequently reported.
Approximately 12% of patients experienced a side effect related to the intravenous infusion; most common were pain at the injection site (6.5%) and local inflammation (3.1%).
The most common adverse reactions associated with treatment in adult women who received IV/Oral ZITHROMAX in trials of pelvic inflammatory disease were related to the gastrointestinal system. Diarrhea (8.5%) and nausea (6.6%) were most commonly reported, followed by vaginitis (2.8%), abdominal pain (1.9%), anorexia (1.9%), rash and pruritus (1.9%). When azithromycin was co-administered with metronidazole in these trials, a higher proportion of women experienced adverse reactions of nausea (10.3%), abdominal pain (3.7%), vomiting (2.8%), infusion site reaction, stomatitis, dizziness, or dyspnea (all at 1.9%).
Adverse reactions that occurred with a frequency of 1% or less included the following:
Gastrointestinal: Dyspepsia, flatulence, mucositis, oral moniliasis, and gastritis.
Nervous system: Headache, somnolence.
Allergic: Bronchospasm.
Special senses: Taste perversion.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of azithromycin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Adverse reactions reported with azithromycin during the postmarketing period in adult and/or pediatric patients for which a causal relationship may not be established include:
Allergic: Arthralgia, edema, urticaria and angioedema.
Cardiovascular: Arrhythmias including ventricular tachycardia and hypotension. There have been reports of QT prolongation, torsades de pointes, and cardiovascular death.
Gastrointestinal: Anorexia, constipation, dyspepsia, flatulence, vomiting/diarrhea, pseudomembranous colitis, pancreatitis, oral candidiasis, pyloric stenosis, and reports of tongue discoloration.
General: Asthenia, paresthesia, fatigue, malaise and anaphylaxis (including fatalities).
Genitourinary: Interstitial nephritis and acute renal failure and vaginitis.
Hematopoietic: Thrombocytopenia.
Liver/biliary: Abnormal liver function, hepatitis, cholestatic jaundice, hepatic necrosis, and hepatic failure. [see Warnings and Precautions (5.2)]
Nervous system: Convulsions, dizziness/vertigo, headache, somnolence, hyperactivity, nervousness, agitation and syncope.
Psychiatric: Aggressive reaction and anxiety.
Skin/appendages: Pruritus, serious skin reactions including, erythema multiforme, AGEP, Stevens-Johnson syndrome, toxic epidermal necrolysis, and DRESS.
Special senses: Hearing disturbances including hearing loss, deafness and/or tinnitus and reports of taste/smell perversion and/or loss.
6.3 Laboratory Abnormalities
Significant abnormalities (irrespective of drug relationship) occurring during the clinical trials were reported as follows:
- elevated ALT (SGPT), AST (SGOT), creatinine (4 to 6%)
- elevated LDH, bilirubin (1 to 3%)
- leukopenia, neutropenia, decreased platelet count, and elevated serum alkaline phosphatase (less than 1%)
When follow-up was provided, changes in laboratory tests appeared to be reversible.
In multiple-dose clinical trials involving more than 750 patients treated with ZITHROMAX (IV/Oral), less than 2% of patients discontinued azithromycin therapy because of treatment-related liver enzyme abnormalities.
Drug Interactions
7 DRUG INTERACTIONS
7.1 Nelfinavir
Co-administration of nelfinavir at steady-state with a single oral dose of azithromycin resulted in increased azithromycin serum concentrations. Although a dose adjustment of azithromycin is not recommended when administered in combination with nelfinavir, close monitoring for known adverse reactions of azithromycin, such as liver enzyme abnormalities and hearing impairment, is warranted. [see Adverse Reactions (6)]
7.2 Warfarin
Spontaneous postmarketing reports suggest that concomitant administration of azithromycin may potentiate the effects of oral anticoagulants such as warfarin, although the prothrombin time was not affected in the dedicated drug interaction study with azithromycin and warfarin. Prothrombin times should be carefully monitored while patients are receiving azithromycin and oral anticoagulants concomitantly.
7.3 Potential Drug-Drug Interaction with Macrolides
Interactions with digoxin, colchicine or phenytoin have not been reported in clinical trials with azithromycin. No specific drug interaction studies have been performed to evaluate potential drug-drug interaction. However, drug interactions have been observed with other macrolide products. Until further data are developed regarding drug interactions when digoxin, colchicine or phenytoin are used with azithromycin careful monitoring of patients is advised.
Use in Specific Populations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from published literature and postmarketing experience over several decades with azithromycin use in pregnant women have not identified any drug-associated risks for major birth defects, miscarriage, or adverse maternal or fetal outcomes (see Data).
Developmental toxicity studies with azithromycin in rats, mice, and rabbits showed no drug-induced fetal malformations at doses up to 4, 2, and 2 times, respectively, an adult human daily dose of 500 mg based on body surface area. Decreased viability and delayed development were observed in the offspring of pregnant rats administered azithromycin from day 6 of pregnancy through weaning at a dose equivalent to 4 times an adult human daily dose of 500 mg based on body surface area (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Human Data
Available data from published observational studies, case series, and case reports over several decades do not suggest an increased risk for major birth defects, miscarriage, or adverse maternal or fetal outcomes with azithromycin use in pregnant women. Limitations of these data include the lack of randomization and inability to control for confounders such as underlying maternal disease and maternal use of concomitant medications.
Animal Data
Reproductive and developmental toxicology studies have not been conducted using IV administration of azithromycin to animals. Azithromycin administered during the period of organogenesis did not cause fetal malformations in rats and mice at oral doses up to 200 mg/kg/day (moderately maternally toxic). Based on body surface area, this dose is approximately 4 (rats) and 2 (mice) times an adult human daily dose of 500 mg. In rabbits administered azithromycin at oral doses of 10, 20, and 40 mg/kg/day during organogenesis, reduced maternal body weight and food consumption were observed in all groups; no evidence of fetotoxicity or teratogenicity was observed at these doses, the highest of which is estimated to be 2 times an adult human daily dose of 500 mg based on body surface area.
In a pre- and postnatal development study, azithromycin was administered orally to pregnant rats from day 6 of pregnancy until weaning at doses of 50 or 200 mg/kg/day. Maternal toxicity (reduced food consumption and body weight gain; increased stress at parturition) was observed at the higher dose. Effects in the offspring were noted at 200 mg/kg/day during the postnatal development period (decreased viability, delayed developmental landmarks). These effects were not observed in a pre- and postnatal rat study when up to 200 mg/kg/day of azithromycin was given orally beginning on day 15 of pregnancy until weaning.
8.2 Lactation
Risk Summary
Azithromycin is present in human milk (see Data). Non-serious adverse reactions have been reported in breastfed infants after maternal administration of azithromycin (see Clinical Considerations). There are no available data on the effects of azithromycin on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ZITHROMAX and any potential adverse effects on the breastfed infant from ZITHROMAX or from the underlying maternal condition.
Clinical Considerations
Advise women to monitor the breastfed infant for diarrhea, vomiting, or rash.
Data
Azithromycin breastmilk concentrations were measured in 20 women after receiving a single 2 g oral dose of azithromycin during labor. Breastmilk samples collected on days 3 and 6 postpartum as well as 2 and 4 weeks postpartum revealed the presence of azithromycin in breastmilk up to 4 weeks after dosing. In another study, a single dose of azithromycin 500 mg was administered intravenously to 8 women prior to incision for cesarean section. Breastmilk (colostrum) samples obtained between 12 and 48 hours after dosing revealed that azithromycin persisted in breastmilk up to 48 hours.
8.4 Pediatric Use
Safety and effectiveness of azithromycin for injection in children or adolescents under 16 years have not been established. In controlled clinical studies, azithromycin has been administered to pediatric patients (age 6 months to 16 years) by the oral route. For information regarding the use of ZITHROMAX (azithromycin for oral suspension) in the treatment of pediatric patients, [see Indications and Usage (1), and Dosage and Administration (2)] of the prescribing information for ZITHROMAX (azithromycin for oral suspension) 100 mg/5 mL and 200 mg/5 mL bottles.
8.5 Geriatric Use
Pharmacokinetic studies with intravenous azithromycin have not been performed in older volunteers. Pharmacokinetics of azithromycin following oral administration in older volunteers (65–85 years old) were similar to those in younger volunteers (18–40 years old) for the 5-day therapeutic regimen.
In multiple-dose clinical trials of intravenous azithromycin in the treatment of community-acquired pneumonia, 45% of patients (188/414) were at least 65 years of age and 22% of patients (91/414) were at least 75 years of age. No overall differences in safety were observed between these subjects and younger subjects in terms of adverse reactions, laboratory abnormalities, and discontinuations. Similar decreases in clinical response were noted in azithromycin- and comparator-treated patients with increasing age.
ZITHROMAX (azithromycin for injection) contains 114 mg (4.96 mEq) of sodium per vial. At the usual recommended doses, patients would receive 114 mg (4.96 mEq) of sodium. The geriatric population may respond with a blunted natriuresis to salt loading. The total sodium content from dietary and non-dietary sources may be clinically important with regard to such diseases as congestive heart failure.
Elderly patients may be more susceptible to development of torsades de pointes arrhythmias than younger patients. [see Warnings and Precautions (5.4)]
Overdosage
Description
11 DESCRIPTION
ZITHROMAX for injection contains the active ingredient azithromycin, an azalide, a subclass of macrolide antibacterial drug, for intravenous injection. Azithromycin has the chemical name (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13- [(2,6-dideoxy-3-C-methyl-3-O -methyl-α-L-ribo-hexopyranosyl)oxy]-2-ethyl-3,4,10-trihydroxy-3,5,6,8,10,12,14-hepta-methyl- 11- [[3,4,6-trideoxy-3-(dimethylamino)-β-D-xylo-hexopyranosyl]oxy]-1-oxa- 6-azacyclopentadecan-15-one. Azithromycin is derived from erythromycin; however, it differs chemically from erythromycin in that a methyl-substituted nitrogen atom is incorporated into the lactone ring. Its molecular formula is C38H72N2O12, and its molecular weight is 749. Azithromycin has the following structural formula:
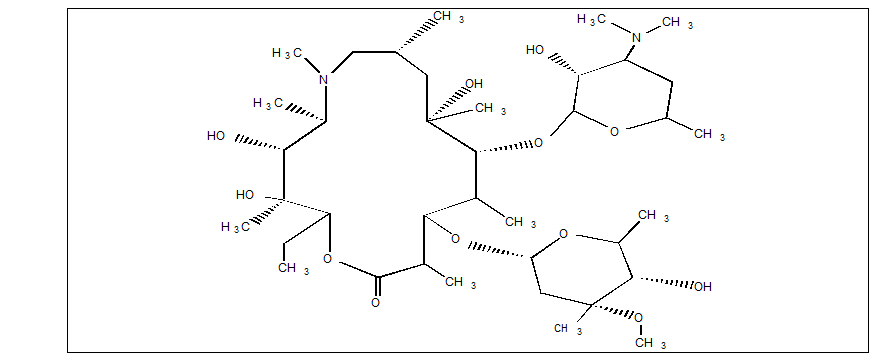
Azithromycin, as the dihydrate, is a white crystalline powder with a molecular formula of C38H72N2O12∙ 2H2O and a molecular weight of 785.
ZITHROMAX for injection consists of azithromycin dihydrate and the following inactive ingredients: anhydrous citric acid and sodium hydroxide. Sodium hydroxide is added to adjust the pH. ZITHROMAX for injection is supplied as white to off-white lyophilized powder in a single-dose vial for intravenous administration. Each vial contains azithromycin dihydrate equivalent to 500 mg of azithromycin, 384.6 mg anhydrous citric acid, and sodium hydroxide. Reconstitution, according to label directions, results in approximately 5 mL of ZITHROMAX for intravenous injection with each mL containing azithromycin dihydrate equivalent to 100 mg of azithromycin, 76.9 mg anhydrous citric acid, and sodium hydroxide.
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
Based on animal models of infection, the antibacterial activity of azithromycin appears to correlate with the ratio of area under the concentration-time curve to minimum inhibitory concentration (AUC/MIC) for certain pathogens (S. pneumoniae and S. aureus). The principal pharmacokinetic/pharmacodynamic parameter best associated with clinical and microbiological cure has not been elucidated in clinical trials with azithromycin.
Cardiac Electrophysiology
QTc interval prolongation was studied in a randomized, placebo-controlled parallel trial in 116 healthy subjects who received either chloroquine (1,000 mg) alone or in combination with oral azithromycin (500 mg, 1,000 mg, and 1,500 mg once daily). Co-administration of azithromycin increased the QTc interval in a dose- and concentration- dependent manner. In comparison to chloroquine alone, the maximum mean (95% upper confidence bound) increases in QTcF were 5 (10) ms, 7 (12) ms and 9 (14) ms with the co-administration of 500 mg, 1,000 mg and 1,500 mg azithromycin, respectively.
Since the mean Cmax of azithromycin following a 500 mg IV dose given over 1 hr is higher than the mean Cmax of azithromycin following the administration of a 1,500 mg oral dose, it is possible that QTc may be prolonged to a greater extent with IV azithromycin at close proximity to a one hour infusion of 500 mg.
12.3 Pharmacokinetics
In patients hospitalized with community-acquired pneumonia receiving single daily one-hour intravenous infusions for 2 to 5 days of 500 mg azithromycin at a concentration of 2 mg/mL, the mean Cmax ± S.D. achieved was 3.63 ± 1.60 mcg/mL, while the 24-hour trough level was 0.20 ± 0.15 mcg/mL, and the AUC24 was 9.60 ± 4.80 mcg∙hr/mL.
The mean Cmax, 24-hour trough and AUC24 values were 1.14 ± 0.14 mcg/mL, 0.18 ± 0.02 mcg/mL, and 8.03 ±0.86 mcg∙hr/mL, respectively, in normal volunteers receiving a 3-hour intravenous infusion of 500 mg azithromycin at a concentration of 1 mg/mL. Similar pharmacokinetic values were obtained in patients hospitalized with community-acquired pneumonia who received the same 3-hour dosage regimen for 2–5 days.
| Infusion Concentration, Duration | Time after starting the infusion (hr) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.5 | 1 | 2 | 3 | 4 | 6 | 8 | 12 | 24 | |
| 2 mg/mL, 1 hr* | 2.98 ±1.12 | 3.63 ±1.73 | 0.60 ±0.31 | 0.40 ±0.23 | 0.33 ±0.16 | 0.26 ±0.14 | 0.27±0.15 | 0.20 ±0.12 | 0.20 ±0.15 |
| 1 mg/mL, 3 hr† | 0.91 ±0.13 | 1.02 ±0.11 | 1.14 ±0.13 | 1.13 ±0.16 | 0.32 ±0.05 | 0.28 ±0.04 | 0.27±0.03 | 0.22 ±0.02 | 0.18 ±0.02 |
Comparison of the plasma pharmacokinetic parameters following the 1st and 5th daily doses of 500 mg intravenous azithromycin showed only an 8% increase in Cmax but a 61% increase in AUC24 reflecting a threefold rise in C24 trough levels.
Following single-oral doses of 500 mg azithromycin (two 250 mg capsules) to 12 healthy volunteers, Cmax, trough level, and AUC24 were reported to be 0.41 mcg/mL, 0.05 mcg/mL, and 2.6 mcg∙hr/mL, respectively. These oral values are approximately 38%, 83%, and 52% of the values observed following a single 500-mg I.V. 3-hour infusion (Cmax: 1.08 mcg/mL, trough: 0.06 mcg/mL, and AUC24: 5 mcg∙hr/mL). Thus, plasma concentrations are higher following the intravenous regimen throughout the 24-hour interval.
Distribution
The serum protein binding of azithromycin is variable in the concentration range approximating human exposure, decreasing from 51% at 0.02 mcg/mL to 7% at 2 mcg/mL.
Tissue concentrations have not been obtained following intravenous infusions of azithromycin, but following oral administration in humans azithromycin has been shown to penetrate into tissues, including skin, lung, tonsil, and cervix.
Tissue levels were determined following a single oral dose of 500 mg azithromycin in 7 gynecological patients. Approximately 17 hr after dosing, azithromycin concentrations were 2.7 mcg/g in ovarian tissue, 3.5 mcg/g in uterine tissue, and 3.3 mcg/g in salpinx. Following a regimen of 500 mg on the first day followed by 250 mg daily for 4 days, concentrations in the cerebrospinal fluid were less than 0.01 mcg/mL in the presence of non-inflamed meninges.
Metabolism
In vitro and in vivo studies to assess the metabolism of azithromycin have not been performed.
Elimination
Plasma concentrations of azithromycin following single 500 mg oral and IV doses declined in a polyphasic pattern with a mean apparent plasma clearance of 630 mL/min and terminal elimination half-life of 68 hr. The prolonged terminal half-life is thought to be due to extensive uptake and subsequent release of drug from tissues.
In a multiple-dose study in 12 normal volunteers utilizing a 500 mg (1 mg/mL) one-hour intravenous-dosage regimen for five days, the amount of administered azithromycin dose excreted in urine in 24 hr was about 11% after the 1st dose and 14% after the 5th dose. These values are greater than the reported 6% excreted unchanged in urine after oral administration of azithromycin. Biliary excretion is a major route of elimination for unchanged drug, following oral administration.
Specific Populations
Patients with Renal Impairment
Azithromycin pharmacokinetics were investigated in 42 adults (21 to 85 years of age) with varying degrees of renal impairment. Following the oral administration of a single 1,000 mg dose of azithromycin, mean Cmax and AUC0–120 increased by 5.1% and 4.2%, respectively in subjects with mild to moderate renal impairment (GFR 10 to 80 mL/min) compared to subjects with normal renal function (GFR >80 mL/min). The mean Cmax and AUC0–120 increased 61% and 35%, respectively in subjects with severe renal impairment (GFR <10 mL/min) compared to subjects with normal renal function (GFR >80 mL/min).
Patients with Hepatic Impairment
The pharmacokinetics of azithromycin in subjects with hepatic impairment has not been established.
Male and Female Patients
There are no significant differences in the disposition of azithromycin between male and female subjects. No dosage adjustment is recommended based on gender.
Geriatric Patients
Pharmacokinetic studies with intravenous azithromycin have not been performed in older volunteers. Pharmacokinetics of azithromycin following oral administration in older volunteers (65–85 years old) were similar to those in younger volunteers (18–40 years old) for the 5-day therapeutic regimen. [see Geriatric Use 8.5)].
Drug Interaction Studies
Drug interaction studies were performed with oral azithromycin and other drugs likely to be co-administered. The effects of co-administration of azithromycin on the pharmacokinetics of other drugs are shown in Table 1 and the effects of other drugs on the pharmacokinetics of azithromycin are shown in Table 2.
Co-administration of azithromycin at therapeutic doses had a modest effect on the pharmacokinetics of the drugs listed in Table 1. No dosage adjustment of drugs listed in Table 1 is recommended when co-administered with azithromycin.
Co-administration of azithromycin with efavirenz or fluconazole had a modest effect on the pharmacokinetics of azithromycin. Nelfinavir significantly increased the Cmax and AUC of azithromycin. No dosage adjustment of azithromycin is recommended when administered with drugs listed in Table 2 [see Drug Interactions (7.3)].
| Co-administered Drug | Dose of Co-administered Drug | Dose of Azithromycin | n | Ratio (with/without azithromycin) of Co-administered Drug Pharmacokinetic Parameters (90% CI); No Effect = 1 | |
|---|---|---|---|---|---|
| Mean Cmax | Mean AUC | ||||
| |||||
| Atorvastatin | 10 mg/day for 8 days | 500 mg/day orally on days 6–8 | 12 | 0.83 (0.63 to 1.08) | 1.01 (0.81 to 1.25) |
| Carbamazepine | 200 mg/day for 2 days, then 200 mg twice a day for 18 days | 500 mg/day orally for days 16–18 | 7 | 0.97 (0.88 to 1.06) | 0.96 (0.88 to 1.06) |
| Cetirizine | 20 mg/day for 11 days | 500 mg orally on day 7, then 250 mg/day on days 8–11 | 14 | 1.03 (0.93 to 1.14) | 1.02 (0.92 to 1.13) |
| Didanosine | 200 mg orally twice a day for 21 days | 1,200 mg/day orally on days 8–21 | 6 | 1.44 (0.85 to 2.43) | 1.14 (0.83 to 1.57) |
| Efavirenz | 400 mg/day for 7 days | 600 mg orally on day 7 | 14 | 1.04* | 0.95* |
| Fluconazole | 200 mg orally single dose | 1,200 mg orally single dose | 18 | 1.04 (0.98 to 1.11) | 1.01 (0.97 to 1.05) |
| Indinavir | 800 mg three times a day for 5 days | 1,200 mg orally on day 5 | 18 | 0.96 (0.86 to 1.08) | 0.9 (0.81 to 1) |
| Midazolam | 15 mg orally on day 3 | 500 mg/day orally for 3 days | 12 | 1.27 (0.89 to 1.81) | 1.26 (1.01 to 1.56) |
| Nelfinavir | 750 mg three times a day for 11 days | 1,200 mg orally on day 9 | 14 | 0.9 (0.81 to 1.01) | 0.85 (0.78 to 0.93) |
| Sildenafil | 100 mg on days 1 and 4 | 500 mg/day orally for 3 days | 12 | 1.16 (0.86 to 1.57) | 0.92 (0.75 to 1.12) |
| Theophylline | 4 mg/kg IV on days 1, 11, 25 | 500 mg orally on day 7, 250 mg/day on days 8–11 | 10 | 1.19 (1.02 to 1.4) | 1.02 (0.86 to 1.22) |
| Theophylline | 300 mg orally BID ×15 days | 500 mg orally on day 6, then 250 mg/day on days 7–10 | 8 | 1.09 (0.92 to 1.29) | 1.08 (0.89 to 1.31) |
| Triazolam | 0.125 mg on day 2 | 500 mg orally on day 1, then 250 mg/day on day 2 | 12 | 1.06* | 1.02* |
| Trimethoprim/Sulfamethoxazole | 160 mg/800 mg/day orally for 7 days | 1,200 mg orally on day 7 | 12 | 0.85 (0.75 to 0.97)/ 0.9 (0.78 to 1.03) | 0.87 (0.8 to 0.95/ 0.96 (0.88 to 1.03) |
| Zidovudine | 500 mg/day orally for 21 days | 600 mg/day orally for 14 days | 5 | 1.12 (0.42 to 3.02) | 0.94 (0.52 to 1.7) |
| Zidovudine | 500 mg/day orally for 21 days | 1,200 mg/day orally for 14 days | 4 | 1.31 (0.43 to 3.97) | 1.3 (0.69 to 2.43) |
| Co-administered Drug | Dose of Co-administered Drug | Dose of Azithromycin | n | Ratio (with/without co-administered drug) of Azithromycin Pharmacokinetic Parameters (90% CI); No Effect = 1 | |
|---|---|---|---|---|---|
| Mean Cmax | Mean AUC | ||||
| |||||
| Efavirenz | 400 mg/day for 7 days | 600 mg orally on day 7 | 14 | 1.22 (1.04 to 1.42) | 0.92* |
| Fluconazole | 200 mg orally single dose | 1,200 mg orally single dose | 18 | 0.82 (0.66 to 1.02) | 1.07 (0.94 to 1.22) |
| Nelfinavir | 750 mg three times a day for 11 days | 1,200 mg orally on day 9 | 14 | 2.36 (1.77 to 3.15) | 2.12 (1.80 to 2.5) |
12.4 Microbiology
Mechanism of Action
Azithromycin acts by binding to the 23S rRNA of the 50S ribosomal subunit of susceptible microorganisms inhibiting bacterial protein synthesis and impeding the assembly of the 50S ribosomal subunit.
Resistance
Azithromycin demonstrates cross-resistance with erythromycin. The most frequently encountered mechanism of resistance to azithromycin is modification of the 23S rRNA target, most often by methylation. Ribosomal modifications can determine cross resistance to other macrolides, lincosamides and streptogramin B (MLSB phenotype).
Antimicrobial Activity
Azithromycin has been shown to be active against the following microorganisms, both in vitro and in clinical infections. [see Indications and Usage (1)]
Gram-positive Bacteria
Staphylococcus aureus
Streptococcus pneumoniae
Gram-negative Bacteria
Haemophilus influenzae
Moraxella catarrhalis
Neisseria gonorrhoeae
Legionella pneumophila
Other Bacteria
Chlamydophila pneumoniae
Chlamydia trachomatis
Mycoplasma hominis
Mycoplasma pneumoniae
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for azithromycin against isolates of similar genus or organism group. However, the efficacy of azithromycin in treating clinical infections caused by these bacteria has not been established in adequate and well-controlled clinical trials.
Aerobic Gram-Positive Bacteria
Streptococci (Groups C, F, G)
Viridans group streptococci
Gram-Negative Bacteria
Bordetella pertussis
Anaerobic Bacteria
Peptostreptococcus species
Prevotella bivia
Other Bacteria
Ureaplasma urealyticum
Nonclinical Toxicology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies in animals have not been performed to evaluate carcinogenic potential. Azithromycin has shown no mutagenic potential in standard laboratory tests: mouse lymphoma assay, human lymphocyte clastogenic assay, and mouse bone marrow clastogenic assay. In fertility studies conducted in male and female rats, oral administration of azithromycin for 64 to 66 days (males) or 15 days (females) prior to and during cohabitation resulted in decreased pregnancy rate at 20 and 30 mg/kg/day when both males and females were treated with azithromycin. This minimal effect on pregnancy rate (approximately 12% reduction compared to concurrent controls) did not become more pronounced when the dose was increased from 20 to 30 mg/kg/day (approximately 0.4 to 0.6 times the adult daily dose of 500 mg based on body surface area) and it was not observed when only one animal in the mated pair was treated. There were no effects on any other reproductive parameters, and there were no effects on fertility at 10 mg/kg/day. The relevance of these findings to patients being treated with azithromycin at the doses and durations recommended in the prescribing information is uncertain.
13.2 Animal Toxicology and/or Pharmacology
Phospholipidosis (intracellular phospholipid accumulation) has been observed in some tissues of mice, rats, and dogs given multiple oral doses of azithromycin. It has been demonstrated in numerous organ systems (e.g., eye, dorsal root ganglia, liver, gallbladder, kidney, spleen, and/or pancreas) in dogs and rats treated with azithromycin at doses which, expressed on the basis of body surface area, are similar to or less than the highest recommended adult human dose. This effect has been shown to be reversible after cessation of azithromycin treatment. Based on the pharmacokinetic data, phospholipidosis has been seen in the rat (50 mg/kg/day dose) at the observed maximal plasma concentration of 1.3 mcg/mL (1.6 times the observed Cmax of 0.821 mcg /mL at the adult dose of 2 g.) Similarly, it has been shown in the dog (10 mg/kg/day dose) at the observed maximal serum concentration of 1 mcg /mL (1.2 times the observed Cmax of 0.821 mcg /mL at the adult dose of 2 g).
Phospholipidosis was also observed in neonatal rats dosed for 18 days at 30 mg/kg/day, which is less than the pediatric dose of 60 mg/kg based on body surface area. It was not observed in neonatal rats treated for 10 days at 40 mg/kg/day with mean maximal serum concentrations of 1.86 mcg/mL, approximately 1.5 times the Cmax of 1.27 mcg/mL at the pediatric dose. Phospholipidosis has been observed in neonatal dogs (10 mg/kg/day) at maximum mean whole blood concentrations of 3.54 mcg/mL, approximately 3 times the pediatric dose Cmax. The significance of the findings for animals and for humans is unknown.
Clinical Studies
14 CLINICAL STUDIES
14.1 Community-Acquired Pneumonia
In a controlled trial of community-acquired pneumonia performed in the U.S., azithromycin (500 mg as a single daily dose by the intravenous route for 2 to 5 days, followed by 500 mg/day by the oral route to complete 7 to 10 days therapy) was compared to cefuroxime (2,250 mg/day in three divided doses by the intravenous route for 2 to 5 days followed by 1,000 mg/day in two divided doses by the oral route to complete 7 to 10 days therapy), with or without erythromycin. For the 291 patients who were evaluable for clinical efficacy, the clinical outcome rates, i.e., cure, improved, and success (cure + improved) among the 277 patients seen at 10 to 14 days post-therapy were as follows:
| Clinical Outcome | Azithromycin | Comparator |
|---|---|---|
| Cure | 46% | 44% |
| Improved | 32% | 30% |
| Success (Cure + Improved) | 78% | 74% |
In a separate, uncontrolled clinical and microbiological trial performed in the U.S., 94 patients with community-acquired pneumonia who received azithromycin in the same regimen were evaluable for clinical efficacy. The clinical outcome rates, i.e., cure, improved, and success (cure + improved) among the 84 patients seen at 10 to 14 days post-therapy were as follows:
| Clinical Outcome | Azithromycin |
|---|---|
| Cure | 60% |
| Improved | 29% |
| Success (Cure + Improved) | 89% |
Microbiological determinations in both trials were made at the pre-treatment visit and, where applicable, were reassessed at later visits. Serological testing was done on baseline and final visit specimens. The following combined presumptive bacteriological eradication rates were obtained from the evaluable groups:
Combined Bacteriological Eradication Rates for Azithromycin:
| (at last completed visit) | Azithromycin |
|---|---|
| |
| S. pneumoniae | 64/67 (96%)* |
| H. influenzae | 41/43 (95%) |
| M. catarrhalis | 9/10 (90%) |
| S. aureus | 9/10 (90%) |
The presumed bacteriological outcomes at 10 to 14 days post-therapy for patients treated with azithromycin with evidence (serology and/or culture) of atypical pathogens for both trials were as follows:
| Evidence of Infection | Total | Cure | Improved | Cure + Improved |
|---|---|---|---|---|
| Mycoplasma pneumoniae | 18 | 11 (61%) | 5 (28%) | 16 (89%) |
| Chlamydia pneumoniae | 34 | 15 (44%) | 13 (38%) | 28 (82%) |
| Legionella pneumophila | 16 | 5 (31%) | 8 (50%) | 13 (81%) |
How Supplied/Storage and Handling
16 HOW SUPPLIED/STORAGE AND HANDLING
ZITHROMAX for injection is supplied as white to off-white lyophilized powder under a vacuum in a single-dose vial equivalent to 500 mg of azithromycin for intravenous administration.
These are packaged as follows:
| Unit of Sale | Concentration |
|---|---|
| NDC 0069-3150-83 | 500 mg/vial |
| Carton containing 10 single-dose vials |
Before reconstitution, store vials at or below 30°C (86°F) [see Dosage and Administration (2.3)].
Patient Counseling Information
17 PATIENT COUNSELING INFORMATION
Patients should be informed of the following serious and potentially serious adverse reactions that have been associated with ZITHROMAX
Diarrhea: Inform patients that diarrhea is a common problem caused by antibacterial drugs which usually ends when the antibacterial is discontinued. Sometimes after starting treatment with antibacterials, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibacterial. If this occurs, patients should notify their physician as soon as possible.
Other

Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Contact Medical Information. 9AM-5PM ET Monday to Friday; excluding holidays.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.