ABRYSVO®
(Respiratory Syncytial Virus Vaccine)
Find ABRYSVO® medical information:
Find ABRYSVO® medical information:
ABRYSVO® Quick Finder
17 PATIENT COUNSELING INFORMATION
17 PATIENT COUNSELING INFORMATION
Prior to administration of this vaccine:
- •
- Inform vaccine recipient of the potential benefits and risks of vaccination with ABRYSVO.
- •
- Advise vaccine recipient to report any adverse events to their healthcare provider or to the Vaccine Adverse Event Reporting System at 1-800-822-7967 and www.vaers.hhs.gov.
This product's labeling may have been updated. For the most recent prescribing information, please visit www.pfizer.com.
Highlights
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ABRYSVO safely and effectively. See full prescribing information for ABRYSVO. ABRYSVO (Respiratory Syncytial Virus Vaccine) for injection, for intramuscular use Initial U.S. Approval: 2023 RECENT MAJOR CHANGESINDICATIONS AND USAGEABRYSVO is a vaccine indicated for
DOSAGE AND ADMINISTRATIONDOSAGE FORMS AND STRENGTHSCONTRAINDICATIONSHistory of severe allergic reaction (e.g., anaphylaxis) to any component of ABRYSVO. (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or VAERS at 1-800-822-7967 or http://vaers.hhs.gov. See 17 for PATIENT COUNSELING INFORMATION. Revised: 3/2024 |
Indications and Usage
1 INDICATIONS AND USAGE
1.1 Immunization of Pregnant Individuals
ABRYSVO is a vaccine indicated for active immunization of pregnant individuals at 32 through 36 weeks gestational age for the prevention of lower respiratory tract disease (LRTD) and severe LRTD caused by respiratory syncytial virus (RSV) in infants from birth through 6 months of age.
Dosage and Administration
2 DOSAGE AND ADMINISTRATION
2.1 Dose and Schedule
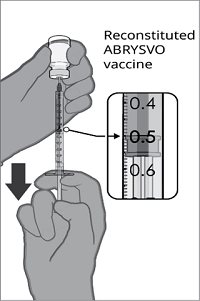
After reconstitution, a single dose of ABRYSVO is either approximately 0.5 mL (vial and prefilled syringe presentation) or 0.5 mL (vial and vial presentation) [see Dosage and Administration (2.2)].
2.2 Presentations and Reconstitution
ABRYSVO is supplied in 2 presentations as follows:
Vial and Prefilled Syringe Presentation
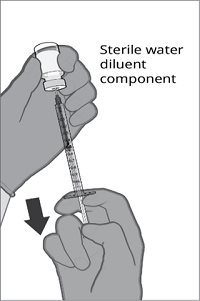
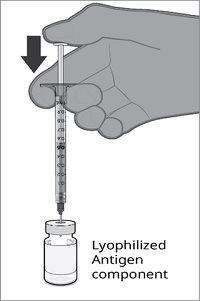
The vial and prefilled syringe presentation is supplied in a kit that includes a vial of Lyophilized Antigen Component (a sterile white powder), a prefilled syringe containing the Sterile Water Diluent Component, and a vial adapter.
Vial and Vial Presentation
The vial and vial presentation is supplied in cartons that include vials of Lyophilized Antigen Component (a sterile white powder) and vials containing the Sterile Water Diluent Component.
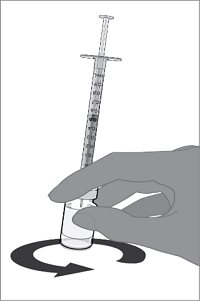
For both presentations, reconstitute the Lyophilized Antigen Component with the accompanying Sterile Water Diluent Component to form ABRYSVO, as described in the instructions below.
Reconstitution Instructions for Vial and Prefilled Syringe Presentation
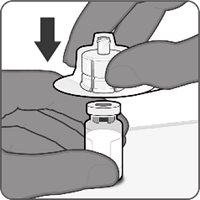
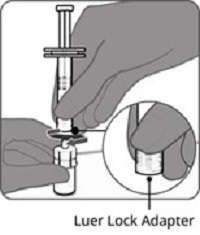
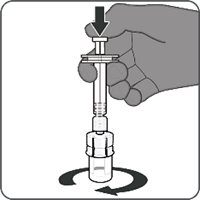
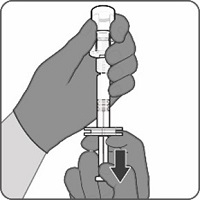
Reconstitution Instructions for the Vial and Vial Presentation
2.3 Administration
For intramuscular injection
After reconstitution, ABRYSVO is a clear and colorless solution. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Discard if either condition is present.
Administer ABRYSVO immediately or store at room temperature [15°C to 30°C (59°F to 86°F)] and use within 4 hours. Discard reconstituted vaccine if not used within 4 hours.
Dosage Forms and Strengths
Contraindications
4 CONTRAINDICATIONS
Do not administer ABRYSVO to individuals with a history of a severe allergic reaction (e.g., anaphylaxis) to any component of ABRYSVO [see Description (11)].
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Potential Risk of Preterm Birth
A numerical imbalance in preterm births in ABRYSVO recipients was observed compared to placebo recipients in two clinical studies [see Adverse Reactions 6.1]. Available data are insufficient to establish or exclude a causal relationship between preterm birth and ABRYSVO. To avoid the potential risk of preterm birth with use of ABRYSVO before 32 weeks of gestation, administer ABRYSVO as indicated in pregnant individuals at 32 through 36 weeks gestational age. Pregnant individuals who were at increased risk of preterm birth were generally excluded from clinical studies of ABRYSVO.
5.2 Management of Acute Allergic Reactions
Appropriate medical treatment used to manage immediate allergic reactions must be immediately available in the event an anaphylactic reaction occurs following administration of ABRYSVO.
5.3 Syncope
Syncope (fainting) may occur in association with administration of injectable vaccines, including ABRYSVO. Procedures should be in place to avoid injury from fainting.
Adverse Reactions
6 ADVERSE REACTIONS
In pregnant individuals, the most commonly reported (≥10%) adverse reactions were pain at the injection site (40.6%), headache (31.0%), muscle pain (26.5%), and nausea (20.0%).
In individuals 60 years of age and older, the most commonly reported (≥10%) adverse reactions were fatigue (15.5%), headache (12.8%), pain at the injection site (10.5%), and muscle pain (10.1%).
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice.
Pregnant Individuals and Infants from Birth Through 6 Months of Age
The safety of ABRYSVO in maternal and infant participants was evaluated in two clinical studies in which approximately 4,000 maternal participants received a single dose of ABRYSVO.
Study 1 (NCT04424316) is an ongoing, Phase 3, randomized, double-blind, multicenter, placebo-controlled study to investigate the efficacy and safety of ABRYSVO administered to pregnant individuals ≤49 years of age with uncomplicated, singleton pregnancies, to protect their infants against RSV disease. Pregnant individuals with high-risk pregnancies were excluded from the study (BMI>40 kg/m2 prior to pregnancy, pregnancies resulting after in vitro fertilization, preeclampsia, eclampsia, uncontrolled gestational hypertension, placental abnormalities, polyhydramnios or oligohydramnios, significant bleeding or blood clotting disorder, unstable endocrine disorders including untreated disorders of glucose intolerance or thyroid disorders). Pregnant individuals with prior pregnancy complications (e.g., history of preterm birth ≤34 weeks gestation, prior stillbirth, neonatal death, previous infant with a known genetic disorder or significant congenital anomaly) could be included, based on the investigators’ judgment, but were generally not enrolled in the study. In this study with 1:1 randomization 3,682 participants received ABRYSVO and 3,675 received placebo (0.5 mL dose, containing the same buffer ingredients in the same quantities as in a single dose of ABRYSVO [see Description (11)]). Infants born in year 1 are to be followed for up to 24 months, and infants born in year 2 will be followed for up to 12 months to assess safety. At the time of data evaluation following a median of 8.9 months (range Day 1-23.8 months), 3,568 infants were born to the maternal participants in the ABRYSVO group and 3,558 in the placebo group, and of these, approximately 45.6% have been followed for 12 months. This multicenter study is being conducted in Argentina, Australia, Brazil, Canada, Chile, Denmark, Finland, Gambia, Japan, Republic of Korea, Mexico, Netherlands, New Zealand, Philippines, South Africa, Spain, Taiwan, and the US.
Demographic characteristics in Study 1 among participants who received ABRYSVO and those who received placebo were generally similar with regard to age, race, and ethnicity. Of the participants in the study, 65% were White, 20% were Black or African American, 13% were Asian, and 29% were Hispanic/Latino. The median maternal age at the time of study vaccination was 29 years (range 16 to 45 years in the ABRYSVO group, 14 to 47 years in the placebo group). The median gestational age at vaccination was 31 weeks and 2 days (range 24-36.9 weeks). ABRYSVO is approved for use for pregnant individuals at 32 through 36 weeks gestational age [see Indications and Usage (1.1)]. The median infant gestational age at birth was 39 weeks and 1 day (range 27 weeks and 3 days to 44 weeks and 2 days). Among the infants born to maternal participants 51% were male and 49% were female.
Study 2 (NCT04032093) was a Phase 2, randomized, placebo-controlled, observer-blinded study that investigated the safety of two dose levels (120 mcg and a higher dose) of ABRYSVO administered to pregnant individuals. ABRYSVO (120 mcg) was administered to 115 maternal participants, and 114 infants were born to these maternal participants. This study was conducted in the US, South Africa, Argentina, and Chile. Demographic characteristics among participants who received ABRYSVO and those who received placebo were generally similar with regard to age, race, and ethnicity. Of the participants in the study, 76% were White, 21% were Black or African American, and 28% were Hispanic/Latino. The median age of participants was 27 years (range 18-42 years). The median gestational age at vaccination was 30 weeks (range 24-36 weeks). ABRYSVO is approved for use for pregnant individuals at 32 through 36 weeks gestational age [see Indications and Usage (1.1)].
For all maternal participants in Study 1, solicited local reactions and systemic events were collected using electronic diaries for 7 days after study vaccination, adverse events for 1 month and obstetric complications, serious adverse events, and adverse events of special interest for the duration of the study. For infant participants, the collection period for nonserious adverse events was from birth to 1 month. Serious adverse events were monitored for at least 1 year for all infant participants and for up to 2 years for half of the infants in Study 1.
Solicited Local and Systemic Reactions in Study 1
The majority of solicited local and systemic reactions in maternal participants resolved within 2-3 days of onset. Severe local reactions were reported for 0.3% of maternal participants in the ABRYSVO group and none in the placebo group, and severe systemic reactions within 7 days after vaccination were reported by 2.3% of maternal participants in both groups.
Solicited local and systemic reactions reported within 7 days after vaccination in Study 1 are presented in Tables 1 and 2.
| ||
Local Reactions | ABRYSVO N=3,663† % | PLACEBO N=3,639† % |
Injection site pain‡ | ||
Any§ | 40.6 | 10.1 |
Mild | 36.1 | 9.3 |
Moderate | 4.4 | 0.9 |
Severe | 0.1 | 0 |
Redness¶ | ||
Any§ | 7.2 | 0.2 |
Mild | 5.0 | 0.1 |
Moderate | 2.1 | 0.1 |
Severe | 0.1 | 0 |
Swelling¶ | ||
Any§ | 6.2 | 0.2 |
Mild | 4.1 | 0.1 |
Moderate | 2.0 | <0.1 |
Severe | <0.1 | 0 |
| ||
Systemic Reactions | ABRYSVO N=3,663† % | PLACEBO N=3,638-3,639† % |
Fever (≥38.0℃) | ||
≥38.0°C | 2.6 | 2.9 |
≥38.0°C to 38.4°C | 1.7 | 1.5 |
>38.5°C to 38.9°C | 0.8 | 1.2 |
>39.0°C to 40.0°C | <0.1 | 0.1 |
>40.0°C | <0.1 | 0.1 |
Fatigue‡ | ||
Any§ | 46.1 | 43.8 |
Mild | 23.4 | 22.8 |
Moderate | 21.4 | 19.6 |
Severe | 1.3 | 1.4 |
Headache‡ | ||
Any§ | 31.0 | 27.6 |
Mild | 20.2 | 17.9 |
Moderate | 10.4 | 9.3 |
Severe | 0.4 | 0.4 |
Muscle pain‡ | ||
Any§ | 26.5 | 17.1 |
Mild | 17.6 | 10.0 |
Moderate | 8.6 | 6.8 |
Severe | 0.4 | 0.3 |
Nausea‡ | ||
Any§ | 20.0 | 19.2 |
Mild | 14.4 | 13.8 |
Moderate | 5.4 | 5.2 |
Severe | 0.2 | 0.2 |
Joint pain‡ | ||
Any§ | 11.6 | 10.5 |
Mild | 6.5 | 6.0 |
Moderate | 4.9 | 4.4 |
Severe | 0.2 | <0.1 |
Diarrhea¶ | ||
Any | 11.2 | 11.5 |
Mild | 9.1 | 9.4 |
Moderate | 2.0 | 1.9 |
Severe | 0.1 | 0.2 |
Vomiting# | ||
Any | 7.8 | 7.0 |
Mild | 6.4 | 5.4 |
Moderate | 1.3 | 1.5 |
Severe | 0.2 | <0.1 |
Unsolicited Adverse Events in Study 1
Unsolicited adverse events reported within 1 month after vaccination by maternal participants were 13.7% in the ABRYSVO group and 13.1% in the placebo group.
The most frequently reported unsolicited adverse events in maternal participants from vaccination through the 1-month follow-up visit were disorders of pregnancy, puerperium and perinatal conditions (7.0% for the ABRYSVO group versus 6.2% for the placebo group).
Serious Adverse Events in Study 1
In Study 1, serious adverse events in maternal participants were reported by 16.2% in the ABRYSVO group and 15.2% in the placebo group occurring any time during the study (see Table 3) with 4.2% serious adverse events in the ABRYSVO group and 3.7% in the placebo group occurring within 1 month after vaccination. Most of the serious adverse events in maternal participants were related to pregnancy complications and occurred after the 1 month period following vaccination.
| ||||
Serious Adverse Reaction | ABRYSVO N=3,682 n (%) | 95% CI | Placebo N=3,675 n (%) | 95% CI |
All Maternal SAEs | 598 (16.2) | (15.1, 17.5) | 558 (15.2) | (14.0, 16.4) |
Pre-eclampsia | 68 (1.8) | (1.4, 2.3) | 53 (1.4) | (1.1, 1.9) |
Gestational hypertension | 41 (1.1) | (0.8, 1.5) | 38 (1.0) | (0.7, 1.4) |
Premature rupture of membranes | 15 (0.4) | (0.2, 0.7) | 16 (0.4) | (0.2, 0.7) |
Preterm premature rupture of membranes | 15 (0.4) | (0.2, 0.7) | 10 (0.3) | (0.1, 0.5) |
Hypertension | 13 (0.4) | (0.2, 0.6) | 6 (0.2) | (0.1, 0.4) |
Maternal death† | 1 (<0.1) | (0.0, 0.2) | 0 | (0.0, 0.1) |
Fetal Death‡ | 10 (0.3) | (0.1, 0.5) | 8 (0.2) | (0.1, 0.4) |
Preterm Births in Study 1 and Study 2
A numerical imbalance in preterm births in ABRYSVO recipients compared to placebo recipients was observed in both Studies 1 and 2. In Study 2, preterm births occurred in 5.3% (6 out of 114) in the ABRYSVO group and 2.6% (3 out of 116) in the placebo group. In the subsequent Study 1, preterm birth events occurred in 5.7% [95% CI: 4.9, 6.5] (202 out of 3,568) in the ABRYSVO group and 4.7% [95% CI: 4.1, 5.5] (169 out of 3,558) in the placebo group. In infants born preterm, 83 infants in the ABRYSVO group and 80 infants in the placebo group remained hospitalized or were readmitted to the hospital in the neonatal period (up to 30 days after birth). Available data are insufficient to establish or exclude a causal relationship between preterm birth and ABRYSVO.
A numerical imbalance in preterm births was also observed in Study 1 among the subgroup of infants born to participants who were vaccinated at 32 through 36 weeks gestation, with 4.2% (68/1,631) in the ABRYSVO group and 3.7% (59/1,610) in the placebo group.
Adverse Reactions in Infants
In Study 1, adverse events in infants from birth to 1 month of age were observed in 37.1% in the ABRYSVO group compared to 34.5% in the placebo group. Low birth weight was observed in 5.1% of participants in the ABRYSVO group versus 4.4% in the placebo group, and neonatal jaundice was observed in 7.2% in the ABRYSVO group versus 6.7% in the placebo group.
Individuals 60 Years of Age and Older
The safety of ABRYSVO was evaluated in Study 3 (NCT05035212) in which 17,215 participants received ABRYSVO and 17,069 received placebo (0.5 mL dose, containing the same buffer ingredients in the same quantities as in a single dose of ABRYSVO [see Description (11)]). Study 3 is an ongoing, multicenter, randomized, double-blind, placebo-controlled study to assess the efficacy and safety of ABRYSVO in individuals 60 years of age and older. This study is being conducted in the US, Argentina, Japan, the Netherlands, Canada, South Africa, and Finland. Demographic characteristics among participants who received ABRYSVO and those who received placebo were generally similar with regard to age, sex, race, and ethnicity. Of the participants in the study, 51% were male and 78% were White, 13% were Black or African American, and 37% were Hispanic/Latino. The median age of participants was 67 years (range 59-97 years).
Solicited local and systemic reactions were collected using electronic diaries for 7 days after study vaccination in 7,169 participants (3,630 ABRYSVO participants and 3,539 placebo recipients) from a subset of sites. For all participants, unsolicited adverse events were collected for one month after study vaccination; serious adverse events (SAEs) are collected throughout study participation.
Solicited Local and Systemic Reactions in Study 3
Solicited local and systemic reactions reported within 7 days after vaccination in Study 3 are presented in Tables 4 and 5.
| Local Reactions | ABRYSVO N=3,619-3,621† % | PLACEBO N=3,532-3,539† % |
|---|---|---|
| ||
Injection site pain‡ | ||
Any§ | 10.5 | 6.0 |
Mild | 9.4 | 5.3 |
Moderate | 1.1 | 0.7 |
Severe | <0.1 | 0 |
Any§ | 2.7 | 0.7 |
Mild | 1.5 | 0.5 |
Moderate | 1.1 | 0.2 |
Severe | 0.1 | 0 |
Any§ | 2.4 | 0.5 |
Mild | 1.5 | 0.2 |
Moderate | 0.9 | 0.2 |
Severe | 0.1 | <0.1 |
| Systemic Reactions | ABRYSVO N=3,619-3,621† % | PLACEBO N=3,532-3,539† % |
|---|---|---|
| ||
Fever (≥38.0℃) | ||
≥38.0°C | 1.4 | 1.4 |
≥38.0°C to 38.4°C | 0.6 | 0.8 |
>38.4°C to 38.9°C | 0.8 | 0.6 |
>38.9°C to 40.0°C | <0.1 | <0.1 |
>40.0°C | 0 | <0.1 |
Fatigue‡ | ||
Any§ | 15.5 | 14.4 |
Mild | 9.3 | 8.4 |
Moderate | 5.9 | 5.8 |
Severe | 0.3 | 0.1 |
Headache‡ | ||
Any§ | 12.8 | 11.7 |
Mild | 9.0 | 8.4 |
Moderate | 3.7 | 3.2 |
Severe | 0.1 | <0.1 |
Muscle pain‡ | ||
Any§ | 10.1 | 8.4 |
Mild | 6.5 | 5.5 |
Moderate | 3.5 | 2.8 |
Severe | 0.2 | <0.1 |
Joint pain‡ | ||
Any§ | 7.5 | 6.9 |
Mild | 4.5 | 3.9 |
Moderate | 2.9 | 2.9 |
Severe | <0.1 | <0.1 |
Nausea‡ | ||
Any§ | 3.4 | 3.7 |
Mild | 2.5 | 3.1 |
Moderate | 0.9 | 0.6 |
Severe | 0 | <0.1 |
Vomiting¶ | ||
Any§ | 0.9 | 0.8 |
Mild | 0.7 | 0.7 |
Moderate | 0.2 | 0.1 |
Severe | 0 | <0.1 |
Diarrhea# | ||
Any§ | 5.9 | 5.2 |
Mild | 4.4 | 4.2 |
Moderate | 1.3 | 0.9 |
Severe | 0.1 | 0.1 |
Solicited local and systemic reactions had a median duration of 1-2 days.
Unsolicited Adverse Events in Study 3
Unsolicited adverse events occurring within 1 month after vaccination were similar between groups, reported in 8.9% and 8.5% of participants who received ABRYSVO and placebo, respectively.
Within 30 days after vaccination, atrial fibrillation was reported in 10 vaccine recipients and 4 placebo recipients (of which 4 in the ABRYSVO group and 3 in the placebo group were serious adverse events); the onset of symptoms was 18 to 30 days post vaccination. The currently available information on atrial fibrillation is insufficient to determine a causal relationship to the vaccine. There were no other notable patterns or numerical imbalances between groups for specific categories of unsolicited adverse events.
Serious Adverse Events in Study 3
In Study 3, SAEs were reported by 2.3% of participants in both the ABRYSVO and placebo groups. Three participants in the ABRYSVO group had SAEs which were assessed as possibly related to study vaccination: Guillain-Barre Syndrome reported 7 days after vaccination, Miller Fisher Syndrome reported 8 days after vaccination, and hypersensitivity reported 8 hours after vaccination.
Drug Interactions
7 DRUG INTERACTIONS
In Study 4 in a concomitant administration study of ABRYSVO and a Tetanus Toxoid, Reduced Diphtheria Toxoid and Acellular Pertussis Vaccine, Adsorbed (Tdap) in non-pregnant women, no safety concerns were identified. Immune responses to RSV A, RSV B, diphtheria, and tetanus were non-inferior to those after separate administration. Lower geometric mean antibody concentrations (GMCs) to the acellular pertussis antigens (pertussis toxin [PT], filamentous hemagglutinin (FHA), and pertactin [PRN]) were measured when ABRYSVO was administered concomitantly with Tdap compared to pertussis GMCs when Tdap was administered alone [see Clinical Studies (14.3)].
Concomitant administration of Tdap with ABRYSVO in pregnant individuals has not been studied.
Use in Specific Populations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in individuals exposed to ABRYSVO during pregnancy. Individuals who received ABRYSVO during pregnancy are encouraged to contact, or have their healthcare provider contact, 1-800-616-3791 to enroll in or obtain information about the registry.
Risk Summary
All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the US general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2% to 4%, and 15% to 20%, respectively, and the estimated background risk of fetal deaths after 20 weeks is 0.6%.
Study 1 enrolled 7,358 pregnant individuals who were randomized 1:1 and received ABRYSVO or placebo (0.5 mL dose, containing the same buffer ingredients in the same quantities as in a single dose of ABRYSVO [see Description (11)]) revealed no evidence for vaccine-associated increase in the risk of congenital anomalies or fetal deaths. Study 2 evaluated 115 pregnant individuals who received ABRYSVO and 117 who received placebo. A numerical imbalance in preterm births in ABRYSVO recipients was observed compared to placebo recipients in these two clinical studies. Available data are insufficient to establish or exclude a causal relationship between preterm birth and ABRYSVO [see Warnings and Precautions (5.1), Adverse Reactions (6.1), Clinical Considerations (8.1), Data (8.1) and Clinical Studies (14.1)].
A developmental toxicity study was performed in female rabbits administered a vaccine formulation containing two times the antigen content of a single human dose of ABRYSVO prior to and during gestation. The study showed no evidence of harm to the fetus or to postnatal survival, growth, or development (see Animal Data).
Clinical Considerations
Maternal Adverse Reactions
In Study 1, 3,682 pregnant individuals received ABRYSVO and 3,676 received placebo. Local and systemic adverse reactions occurred with greater frequency in the ABRYSVO group. Serious adverse reactions observed in pregnant individuals at a higher rate in the ABRYSVO group compared to the placebo group included pre-eclampsia (1.8% versus 1.4%) and gestational hypertension (1.1% versus 1.0%) [see Adverse Reactions (6.1)].
ABRYSVO has not been studied in pregnant individuals less than 24 weeks gestational age, and those at increased risk for preterm birth.
Fetal/Neonatal Adverse Reactions
The infant safety population included 3,568 and 3,558 infants born to individuals in the ABRYSVO or placebo group, respectively. There were 10 (0.3%) fetal deaths in the ABRYSVO group and 8 (0.2%) in the placebo group. Among the infants born to individuals in the ABRYSVO group and in the placebo group, 202 (5.7%) and 169 (4.7%), respectively, were born preterm [see Warnings and Precautions (5.1), Adverse Reactions (6.1) and Clinical Studies (14.1)]. Low birth weight was observed in 5.1% of participants in the ABRYSVO group versus 4.4% in the placebo group, and neonatal jaundice was observed in 7.2% in the ABRYSVO group versus 6.7% in the placebo group. [see Adverse Reactions (6.1)]. For mortality in the neonatal period among infants born to pregnant individuals in Study 1, there were 2 deaths in the ABRYSVO group and 5 in the placebo group, and for overall mortality including after the neonatal period there were 5 deaths in the ABRYSVO group and 12 in the placebo group. Congenital abnormalities were reported in 5.0% in the ABRYSVO group and 6.2% in the placebo group.
Available data are insufficient to establish or exclude a causal relationship between preterm birth and ABRYSVO. To avoid the potential risk of preterm birth with use of ABRYSVO before 32 weeks of gestation, administer ABRYSVO as indicated in pregnant individuals at 32 through 36 weeks gestational age.
Data
Human Data
In Study 1, 3,682 pregnant individuals received ABRYSVO and 3,676 received placebo at 24 through 36 weeks’ gestation. The infant safety population included 3,568 and 3,558 infants born to individuals in the ABRYSVO or placebo group, respectively. Among the infants born to individuals in the ABRYSVO group and in the placebo group, 202 (5.7%) and 169 (4.7%), respectively, had adverse events of preterm birth and 180 (5.0%) and 220 (6.2%), respectively, had reported congenital malformations or anomalies. There were 10 (0.3%) fetal deaths in the ABRYSVO group and 8 (0.2%) in the placebo group.
A pre- and post-natal developmental toxicity study with an embryo-fetal developmental toxicity phase was performed in female New Zealand White rabbits. Rabbits were administered 4 doses by intramuscular injection: at 3 weeks and at 1 week prior to mating, and on gestation days 10 and 24. On each occasion, rabbits received 0.5 mL of a vaccine formulation containing twice the antigen content of F glycoproteins of RSV A and RSV B (120 mcg RSV preF A and 120 mcg RSV preF B), stabilized in prefusion conformation as contained in a single human dose of ABRYSVO [see Description (11)]. No adverse effects on mating, female fertility, or on embryo/fetal or post-natal survival, growth, or development were observed. There were no vaccine-related fetal malformations or variations.
8.2 Lactation
Risk Summary
It is not known whether ABRYSVO is excreted in human milk. Data are not available to assess the effects of ABRYSVO on the breastfed infant or on milk production/excretion. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ABRYSVO and any potential adverse effects on the breastfed child from ABRYSVO or from the underlying maternal condition. For preventative vaccines, the underlying maternal condition is susceptibility to disease prevented by the vaccine.
8.4 Pediatric Use
The safety and effectiveness of ABRYSVO to prevent RSV LRTD and severe RSV LRTD in infants born to individuals vaccinated at younger than 10 years of age have not been established.
The safety and effectiveness of ABRYSVO to prevent RSV LRTD in non-pregnant individuals younger than 18 years of age via active immunization have not been established.
8.5 Geriatric Use
ABRYSVO is approved for use in individuals 60 years of age and older. In Study 3, of the 17,215 recipients who received ABRYSVO 62% (n=10,756) were aged 60-69 years of age, 32% (n=5,488) were 70-79 years of age and 6% (n=970) were ≥80 years of age [see Adverse Reactions (6.1) and Clinical Studies (14.1)].
Description
11 DESCRIPTION
ABRYSVO (Respiratory Syncytial Virus Vaccine) is a sterile solution for intramuscular injection. The vaccine is supplied as a vial of Lyophilized Antigen Component that is reconstituted at the time of use with a Sterile Water Diluent Component. The antigen component contains recombinant RSV preF A and RSV preF B.
The RSV preF A and RSV preF B recombinant proteins are expressed in genetically engineered Chinese Hamster Ovary cell lines grown in suspension culture using chemically-defined media, without antibiotics or animal-derived components. The recombinant proteins are purified through a series of column chromatography and filtration steps followed by formulation, filling into vials, and lyophilization.
After reconstitution, a single dose of ABRYSVO is formulated to contain 120 mcg of RSV stabilized prefusion F proteins (60 mcg RSV preF A and 60 mcg RSV preF B) per 0.5 mL. ABRYSVO also contains the following buffer ingredients: 0.11 mg tromethamine, 1.04 mg tromethamine hydrochloride, 11.3 mg sucrose, 22.5 mg mannitol, 0.08 mg polysorbate 80, and 1.1 mg sodium chloride per 0.5 mL. ABRYSVO is a sterile, clear, and colorless solution.
ABRYSVO contains no preservatives. Each dose may also contain residual amounts of host cell proteins (≤0.1% w/w) and DNA (<0.4 ng/mg of total protein) from the manufacturing process.
The vial stopper and tip cap and rubber plunger of the prefilled syringe are not made with natural rubber latex.
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Active Immunization
ABRYSVO induces an immune response against RSV pre F that protects against lower respiratory tract disease caused by RSV.
Passive Immunization
Antibodies to RSV antigens from individuals vaccinated in pregnancy are transferred transplacentally to protect infants younger than 6 months of age against LRTD and severe LRTD caused by RSV.
Nonclinical Toxicology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
ABRYSVO has not been evaluated for the potential to cause carcinogenicity, genotoxicity, or impairment of male fertility. A developmental toxicity study in female rabbits revealed no evidence of impaired female fertility after administration of a vaccine formulation containing two times the antigen content of a single human dose of ABRYSVO [see Use in Specific Populations (8.1)].
Clinical Studies
14 CLINICAL STUDIES
14.1 Study in Pregnant Individuals for Efficacy in Their Infants from Birth Through 6 Months of Age
Study 1 (NCT04424316) is a Phase 3 study that assessed the efficacy of ABRYSVO in the prevention of RSV-associated lower respiratory tract disease (LRTD) in infants born to individuals vaccinated during pregnancy. The study evaluated the efficacy of ABRYSVO to prevent RSV-associated LRTD and severe RSV-LRTD in infants within 90, 120, 150, and 180 days after birth. Participants were randomized (1:1) to receive ABRYSVO (0.5 mL dose) or placebo (0.5 mL dose containing the same buffer ingredients in the same quantities as in a single dose of ABRYSVO [see Description (11)]). This study includes sites in both the northern and southern hemispheres. Vaccine efficacy (VE) was defined as the relative risk reduction of the endpoints of severe LRTD caused by RSV and LRTD cause by RSV in infants born to individuals who received ABRYSVO compared to infants born to individuals who received placebo. The demographic characteristics of Study 1 are described in Clinical Trials Experience Section 6.1.
Maternal participants were randomized (1:1) to receive ABRYSVO (3,695) or placebo (3,697). RSV-associated LRTD in infants was defined as a medically attended visit with a reverse transcription-polymerase chain reaction (RT-PCR) confirmed RSV illness with one or more of the following respiratory symptoms: tachypnea (respiratory rate ≥60 breaths/minute [<2 months of age], ≥50 breaths/minute [≥2 to 12 months of age], or ≥40 breaths/minute [≥12-24 months of age]); SpO2 measured in room air <95%; chest wall indrawing. RSV-associated severe LRTD was a subset defined as meeting the LRTD RSV criteria plus at least one of the following: tachypnea (respiratory rate ≥70 breaths per minute [<2 months of age], ≥60 breaths per minute [≥2 to 12 months of age], or ≥50 bpm [≥12 to 24 months of age]); SpO2 measured in room air <93%; high-flow nasal cannula or mechanical ventilation (invasive or noninvasive), ICU admission for >4 hours and/or failure to respond/unconscious. Secondary efficacy endpoints included hospitalizations due to RSV.
The VE results met the statistical criterion for success (a CI lower bound >20%) for reducing severe LRTD due to RSV, at all timepoints to within 180 days. The VE results did not meet the statistical criterion for success (a CI lower bound >20%) for reducing LRTD due to RSV; however, clinically meaningful efficacy was observed after 90 days through 180 days after birth.
Vaccine efficacy information is presented in Tables 6 to 10.
| CI - confidence interval; N – number of participants; RSV – respiratory syncytial virus; VE - vaccine efficacy | |||
Time Period | ABRYSVO Number of Cases N=3,495† | PLACEBO Number of Cases N=3,480† | VE (%) (CI)‡ |
90 days | 6 | 33 | 81.8 (40.6, 96.3) |
120 days | 12 | 46 | 73.9 (45.6, 88.8) |
150 days | 16 | 55 | 70.9 (44.5, 85.9) |
180 days | 19 | 62 | 69.4 (44.3, 84.1) |
| CI - confidence interval; N – number of participants; RSV – respiratory syncytial virus; VE - vaccine efficacy | |||
Time Period | ABRYSVO Number of Cases N=3,495† | PLACEBO Number of Cases N=3,480† | VE (%) (CI)‡ |
90 days | 24 | 56 | 57.1 (14.7, 79.8) |
120 days | 35 | 81 | 56.8 (31.2, 73.5) |
150 days | 47 | 99 | 52.5 (28.7, 68.9) |
180 days | 57 | 117 | 51.3 (29.4, 66.8) |
| CI - confidence interval; N – number of participants; n - number of cases; RSV – respiratory syncytial virus; VE - vaccine efficacy | |||
Time Period | ABRYSVO Number of Cases N=1572† | PLACEBO Number of Cases N=1539† | VE (%) (CI)‡ |
90 days | 1 | 11 | 91.1 (38.8, 99.8) |
180 days | 6 | 25 | 76.5 (41.3, 92.1) |
| CI - confidence interval; N – number of participants; n - number of cases; RSV – respiratory syncytial virus; VE - vaccine efficacy | |||
Time Period | ABRYSVO Number of Cases N=1572† | PLACEBO Number of Cases N=1539† | VE (%) (CI)‡ |
90 days | 14 | 21 | 34.7 (-34.6, 69.3) |
180 days | 24 | 55 | 57.3 (29.8, 74.7) |
| CI - confidence interval; N – number of participants; n - number of cases; RSV – respiratory syncytial virus; VE - vaccine efficacy | |||
Time Period | ABRYSVO Number of Cases N=3,495† | PLACEBO Number of Cases N=3,480† | VE (%) (CI)‡ |
90 days | 10 | 31 | 67.7 (15.9, 89.5) |
120 days | 15 | 37 | 59.5 (8.3, 83.7) |
150 days | 17 | 39 | 56.4 (5.2, 81.5) |
180 days | 19 | 44 | 56.8 (10.1, 80.7) |
14.2 Efficacy in Individuals 60 Years of Age and Older
Study 3 (NCT05035212) is an ongoing Phase 3, multicenter, randomized, double-blind, placebo-controlled study to assess the efficacy and safety of ABRYSVO in the prevention of RSV-associated lower respiratory tract disease in individuals 60 years of age and older. Participants are planned to be followed for up to two RSV seasons, approximately 25 months.
Participants were randomized (1:1) to receive ABRYSVO (n=17,197) or placebo (n=17,186). Randomization was stratified by age, 60-69 years (n=21,499, 63%), 70-79 years (n=10,948, 32%), and ≥80 years (n=1,934, 6%). Healthy adults and adults with stable chronic diseases were included. Among enrolled participants 15% had stable chronic cardiopulmonary conditions such as chronic obstructive pulmonary disease (COPD), asthma, or congestive heart failure (CHF).
Starting 14 days after study vaccination (study Day 15), all participants were actively monitored for onset of acute respiratory illness (ARI) symptoms: new or increased sore throat, nasal congestion, nasal discharge, cough, wheezing, sputum production, or shortness of breath. If the participant experienced 1 or more ARI symptoms, a mid-turbinate nasal swab was collected within 7 days of onset of symptoms and tested by reverse transcriptase polymerase chain reaction (RT-PCR) for RSV.
RSV-associated lower respiratory tract disease (RSV-LRTD) was evaluated in Study 3. A case of RSV-LRTD was defined as an RT-PCR confirmed RSV illness with two or more, or three or more, of the following respiratory symptoms within 7 days of symptom onset and lasting more than 1 day during the same illness: new or increased cough, wheezing, sputum production, shortness of breath, or tachypnea (≥25 breaths/min or 15% increase from resting baseline). A case of RSV-associated severe lower respiratory tract disease was defined as a case meeting the RSV-LRTD criteria plus at least one of the following: hospitalization due to RSV-LRTD, new or increased oxygen supplementation, or mechanical ventilation including Continuous Positive Airway Pressure (CPAP).
Efficacy against Respiratory Syncytial Virus-associated Lower Respiratory Tract Disease in Individuals 60 Years of Age and Older
Vaccine efficacy (VE), against RSV-LRTD, defined as the relative risk reduction of first episode of RSV-LRTD in the ABRYSVO group compared to the placebo group in the first RSV season, was assessed.
The study met the pre-specified success criteria for demonstration of efficacy of ABRYSVO for the primary objectives of prevention of RSV-LRTD with ≥2 symptoms and prevention of RSV-LRTD with ≥3 symptoms. The median duration of follow-up for efficacy was 7 months.
Vaccine efficacy information is presented in Table 11.
| CI – confidence interval; N – number of participants; n = number of cases; RSV – respiratory syncytial virus; VE – vaccine efficacy (VE based on case count ratio is calculated as 1-(P/[1-P]), where P is the number of RSVpreF cases divided by the total number of cases) | |||
Efficacy Endpoint | ABRYSVO N=16,306† n | Placebo N=16,308† n | VE (%) (96.66% CI) |
First episode of RSV-associated lower respiratory tract disease with ≥2 symptoms | 11 | 33 | 66.7 (28.8, 85.8) |
First episode of RSV-associated lower respiratory tract disease with ≥3 symptoms | 2 | 14 | 85.7 (32.0, 98.7) |
There were 2 cases of RSV-associated severe lower respiratory tract disease in the placebo group and no cases in the ABRYSVO group.
14.3 Concomitant Vaccine Administration with Tetanus Toxoid, Reduced Diphtheria Toxoid and Acellular Pertussis Vaccine, Adsorbed
Study 4 (NCT04071158) was a Phase 2, placebo-controlled, randomized, observer-blind study to evaluate the safety, tolerability, and immunogenicity of ABRYSVO (at dose levels 120 µg and 240 µg, with or without Al(OH)3) when administered concomitantly with Tdap in non-pregnant women 18 through 49 years of age.
Antibody responses to antigens contained in ABRYSVO and Tdap were assessed 1 month after vaccination in a population of non-pregnant adult individuals. Lower geometric mean antibody concentrations (GMCs) to the acellular pertussis antigens (pertussis toxin [PT], filamentous hemagglutinin (FHA), and pertactin [PRN]) were observed when ABRYSVO was administered concomitantly with a tetanus, diphtheria and acellular pertussis vaccine (Tdap) compared to pertussis GMCs when Tdap was administered alone. The lower limit (LL) of the 2-sided 95% confidence interval of the GMC ratio (GMC ABRYSVO+Tdap /GMC Tdap) was 0.64 for PT, 0.50 for FHA, and 0.48 for PRN, which did not meet the pre-specified non-inferiority criterion (lower limit of the 95% confidence interval for the GMC ratio is >0.67). The clinical relevance of this finding is unknown. The non-inferiority criteria for tetanus, diphtheria and RSV vaccine antigens were met [see Drug Interactions (7)].
How Supplied/Storage and Handling
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
ABRYSVO vial and prefilled syringe presentation is supplied in cartons of 1, 5, and 10 kits, without needles. Each kit includes a vial of Lyophilized Antigen Component (NDC 0069-0207-01), a prefilled syringe containing Sterile Water Diluent Component (NDC 0069-0250-01), and a vial adapter.
Carton: 1 kit | NDC 0069-0344-01 |
Carton: 5 kits | NDC 0069-0344-05 |
Carton: 10 kits | NDC 0069-0344-10 |
ABRYSVO vial and vial presentation is supplied in cartons of 5 and 10 doses packaged without syringes or needles. Each carton includes vials of Lyophilized Antigen Component (NDC 0069-0207-01) and vials of Sterile Water Diluent Component (NDC 0069-0651-01).
Carton: 5 doses | NDC 0069-1265-10 |
Carton: 10 doses | NDC 0069-1265-20 |
The vial stopper and the tip cap and rubber plunger of the prefilled syringe are not made with natural rubber latex.
Patient Counseling Information
17 PATIENT COUNSELING INFORMATION
Prior to administration of this vaccine:
- •
- Inform vaccine recipient of the potential benefits and risks of vaccination with ABRYSVO.
- •
- Advise vaccine recipient to report any adverse events to their healthcare provider or to the Vaccine Adverse Event Reporting System at 1-800-822-7967 and www.vaers.hhs.gov.
This product's labeling may have been updated. For the most recent prescribing information, please visit www.pfizer.com.
Other

Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Contact Medical Information. 9AM-5PM ET Monday to Friday; excluding holidays.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.