XELJANZ / XELJANZ XR Clinical Pharmacology
(tofacitinib)
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tofacitinib is a Janus kinase (JAK) inhibitor. JAKs are intracellular enzymes which transmit signals arising from cytokine or growth factor-receptor interactions on the cellular membrane to influence cellular processes of hematopoiesis and immune cell function. Within the signaling pathway, JAKs phosphorylate and activate Signal Transducers and Activators of Transcription (STATs) which modulate intracellular activity including gene expression. Tofacitinib modulates the signaling pathway at the point of JAKs, preventing the phosphorylation and activation of STATs. JAK enzymes transmit cytokine signaling through pairing of JAKs (e.g., JAK1/JAK3, JAK1/JAK2, JAK1/TyK2, JAK2/JAK2). Tofacitinib inhibited the in vitro activities of JAK1/JAK2, JAK1/JAK3, and JAK2/JAK2 combinations with IC50 of 406, 56, and 1377 nM, respectively. However, the relevance of specific JAK combinations to therapeutic effectiveness is not known.
12.2 Pharmacodynamics
Treatment with XELJANZ was associated with dose-dependent reductions of circulating CD16/56+ natural killer cells, with estimated maximum reductions occurring at approximately 8–10 weeks after initiation of therapy. These changes generally resolved within 2–6 weeks after discontinuation of treatment. Treatment with XELJANZ was associated with dose-dependent increases in B cell counts. Changes in circulating T-lymphocyte counts and T-lymphocyte subsets (CD3+, CD4+ and CD8+) were small and inconsistent. The clinical significance of these changes is unknown.
Total serum IgG, IgM, and IgA levels after 6-month dosing in patients with rheumatoid arthritis were lower than placebo; however, changes were small and not dose-dependent.
After treatment with XELJANZ in patients with rheumatoid arthritis, rapid decreases in serum C-reactive protein (CRP) were observed and maintained throughout dosing. Changes in CRP observed with XELJANZ treatment do not reverse fully within 2 weeks after discontinuation, indicating a longer duration of pharmacodynamic activity compared to the pharmacokinetic half-life.
Similar changes in T cells, B cells, and serum CRP have been observed in patients with active psoriatic arthritis although reversibility was not assessed. Total serum immunoglobulins were not assessed in patients with active psoriatic arthritis.
12.3 Pharmacokinetics
XELJANZ/XELJANZ Oral Solution
Following oral administration of XELJANZ/XELJANZ Oral Solution, peak plasma concentrations are reached within 0.5–1 hour, elimination half-life is about 3 hours and a dose-proportional increase in systemic exposure was observed in the therapeutic dose range. Steady state concentrations are achieved in 24–48 hours with negligible accumulation after twice daily administration.
XELJANZ XR
Following oral administration of XELJANZ XR, peak plasma concentrations are reached at 4 hours and half-life is about 6 to 8 hours. Steady state concentrations are achieved within 48 hours with negligible accumulation after once daily administration.
| PK Parameters* (CV%) | XELJANZ | XELJANZ XR | ||
|---|---|---|---|---|
| Dosing Regimen | 5 mg Twice Daily | 10 mg Twice Daily | 11 mg Once Daily | 22 mg Once Daily |
| Abbreviations: AUC24 = area under the concentration-time profile from time 0 to 24 hours; Cmax = maximum plasma concentration; Cmin = minimum plasma concentration; Tmax = time to Cmax; CV = Coefficient of variation. | ||||
AUC24 (ng.hr/mL) | 263.4 (15) | 539.6 (22) | 269.0 (18) | 596.6 (19) |
Cmax (ng/mL) | 42.7 (26) | 84.7 (18) | 38.2 (15) | 83.8 (25) |
Cmin (ng/mL) | 1.41 (40) | 3.10 (54) | 1.07 (69) | 3.11 (43) |
Tmax (hours) | 1.0 | 0.8 | 4.0 | 4.0 |
Absorption
XELJANZ
The absolute oral bioavailability of XELJANZ is 74%. Coadministration of XELJANZ with a high-fat meal resulted in no changes in AUC while Cmax was reduced by 32%. In clinical trials, XELJANZ was administered without regard to meals [see Dosage and Administration (2.1)].
Distribution
After intravenous administration, the volume of distribution is 87 L. The protein binding of tofacitinib is approximately 40%. Tofacitinib binds predominantly to albumin and does not appear to bind to α1-acid glycoprotein. Tofacitinib distributes equally between red blood cells and plasma.
Metabolism and Excretion
Clearance mechanisms for tofacitinib are approximately 70% hepatic metabolism and 30% renal excretion of the parent drug. The metabolism of tofacitinib is primarily mediated by CYP3A4 with minor contribution from CYP2C19. In a human radiolabeled study, more than 65% of the total circulating radioactivity was accounted for by unchanged tofacitinib, with the remaining 35% attributed to 8 metabolites, each accounting for less than 8% of total radioactivity. The pharmacologic activity of tofacitinib is attributed to the parent molecule.
Pharmacokinetics in Patient Populations
Population pharmacokinetic analyses indicated that pharmacokinetic characteristics were similar between patients with rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and UC. The coefficient of variation (%) in AUC of tofacitinib were generally similar across different disease patients, ranging from 22% to 34% (Table 8).
| Pharmacokinetic Parameters* Geometric Mean (CV%) | XELJANZ 5 mg Twice Daily | XELJANZ 10 mg Twice Daily | |||
|---|---|---|---|---|---|
| Rheumatoid Arthritis | Psoriatic Arthritis | Ankylosing Spondylitis | Ulcerative Colitis | Ulcerative Colitis | |
| Abbreviations: AUC0–24,ss=area under the plasma concentration-time curve over 24 hours at steady state; CV=coefficient of variation. | |||||
| |||||
AUC0–24,ss | 504 | 419 | 381 | 423 | 807 |
Specific Populations
Covariate evaluation as part of population PK analyses in adult patient populations indicated no clinically relevant change in tofacitinib exposure, after accounting for differences in renal function (i.e., creatinine clearance) between patients, based on age, weight, gender and race (Figure 1). An approximately linear relationship between body weight and volume of distribution was observed, resulting in higher peak (Cmax) and lower trough (Cmin) concentrations in lighter patients. However, this difference is not considered to be clinically relevant.
Covariate evaluation as part of population PK analyses in pcJIA patients identified body weight significantly impacting tofacitinib exposure, which supports weight-based dosing in this population. No additional dose adjustment is needed based on age, gender, race, or disease severity in pcJIA patients.
The effect of renal and hepatic impairment and other intrinsic factors on the pharmacokinetics of tofacitinib is shown in Figure 1.
Figure 1: Impact of Intrinsic Factors on Tofacitinib Pharmacokinetics
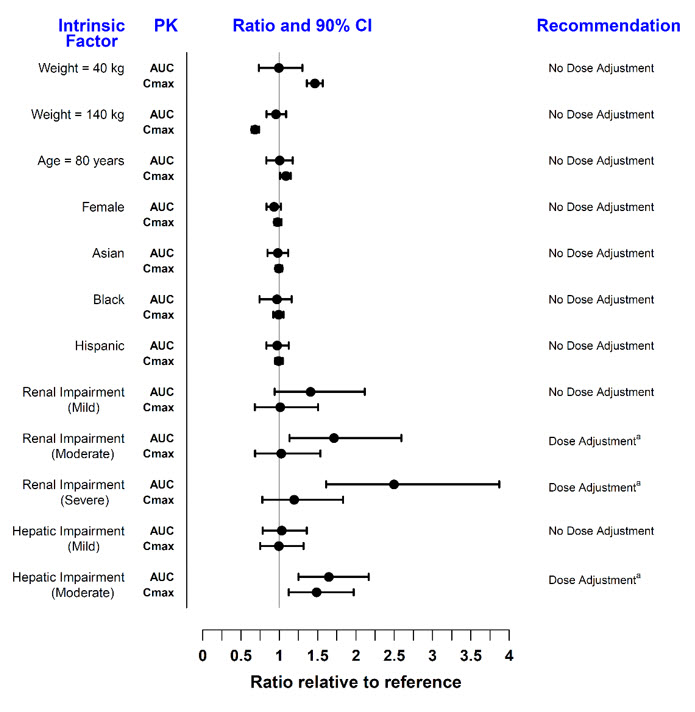
In subjects with ESRD maintained on hemodialysis, mean AUC was approximately 40% higher compared with historical healthy subject data, consistent with approximately 30% contribution of renal clearance to the total clearance of tofacitinib. Dose adjustment is recommended in RA, PsA, AS, UC, and pcJIA patients with ESRD maintained on hemodialysis [see Dosage and Administration (2.2, 2.3, 2.4)].
Drug Interaction Studies
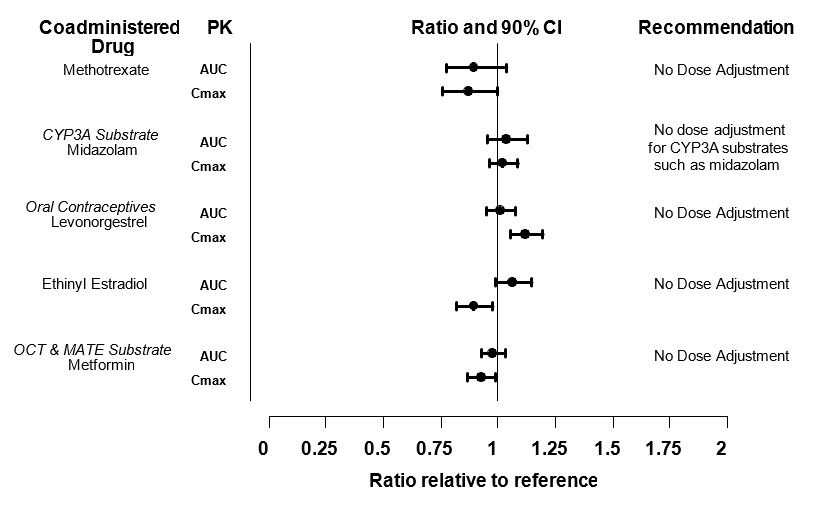
Potential for XELJANZ/XELJANZ XR/XELJANZ Oral Solution to Influence the PK of Other Drugs
In vitro studies indicate that tofacitinib does not significantly inhibit or induce the activity of the major human drug-metabolizing CYPs (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) at concentrations corresponding to the steady state Cmax of a 10 mg twice daily dose. These in vitro results were confirmed by a human drug interaction study showing no changes in the pharmacokinetics of midazolam, a highly sensitive CYP3A4 substrate, when coadministered with XELJANZ.
In vitro studies indicate that tofacitinib does not significantly inhibit the activity of the major human drug-metabolizing uridine 5'-diphospho-glucuronosyltransferases (UGTs) [UGT1A1, UGT1A4, UGT1A6, UGT1A9, and UGT2B7] at concentrations exceeding 250 times the steady state Cmax of a 10 mg twice daily dose.
In rheumatoid arthritis patients, the oral clearance of tofacitinib does not vary with time, indicating that tofacitinib does not normalize CYP enzyme activity in rheumatoid arthritis patients. Therefore, coadministration with XELJANZ/XELJANZ XR is not expected to result in clinically relevant increases in the metabolism of CYP substrates in rheumatoid arthritis patients.
In vitro data indicate that the potential for tofacitinib to inhibit transporters such as P-glycoprotein, organic anionic or cationic transporters at therapeutic concentrations is low.
Dosing recommendations for coadministered drugs following administration with XELJANZ/XELJANZ XR/XELJANZ Oral Solution are shown in Figure 2.
Figure 2: Impact of Tofacitinib on the Pharmacokinetics of Other Drugs
| Note: Reference group is administration of concomitant medication alone; OCT = Organic Cationic Transporter; MATE = Multidrug and Toxic Compound Extrusion. |
 |
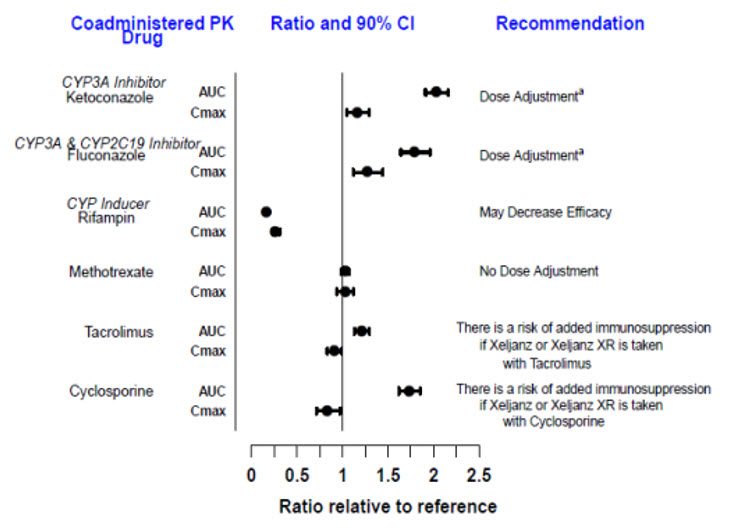
Potential for Other Drugs to Influence the Pharmacokinetics of Tofacitinib
Since tofacitinib is metabolized by CYP3A4, interaction with drugs that inhibit or induce CYP3A4 is likely. Inhibitors of CYP2C19 alone or P-glycoprotein are unlikely to substantially alter the pharmacokinetics of tofacitinib (see Figure 3).
Figure 3: Impact of Other Drugs on the Pharmacokinetics of Tofacitinib
| Note: Reference group is administration of tofacitinib alone. a [see Dosage and Administration (2.2, 2.3, 2.4), Drug Interactions (7)]. |
 |
Find XELJANZ / XELJANZ XR medical information:
Find XELJANZ / XELJANZ XR medical information:
XELJANZ / XELJANZ XR Quick Finder
Health Professional Information
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tofacitinib is a Janus kinase (JAK) inhibitor. JAKs are intracellular enzymes which transmit signals arising from cytokine or growth factor-receptor interactions on the cellular membrane to influence cellular processes of hematopoiesis and immune cell function. Within the signaling pathway, JAKs phosphorylate and activate Signal Transducers and Activators of Transcription (STATs) which modulate intracellular activity including gene expression. Tofacitinib modulates the signaling pathway at the point of JAKs, preventing the phosphorylation and activation of STATs. JAK enzymes transmit cytokine signaling through pairing of JAKs (e.g., JAK1/JAK3, JAK1/JAK2, JAK1/TyK2, JAK2/JAK2). Tofacitinib inhibited the in vitro activities of JAK1/JAK2, JAK1/JAK3, and JAK2/JAK2 combinations with IC50 of 406, 56, and 1377 nM, respectively. However, the relevance of specific JAK combinations to therapeutic effectiveness is not known.
12.2 Pharmacodynamics
Treatment with XELJANZ was associated with dose-dependent reductions of circulating CD16/56+ natural killer cells, with estimated maximum reductions occurring at approximately 8–10 weeks after initiation of therapy. These changes generally resolved within 2–6 weeks after discontinuation of treatment. Treatment with XELJANZ was associated with dose-dependent increases in B cell counts. Changes in circulating T-lymphocyte counts and T-lymphocyte subsets (CD3+, CD4+ and CD8+) were small and inconsistent. The clinical significance of these changes is unknown.
Total serum IgG, IgM, and IgA levels after 6-month dosing in patients with rheumatoid arthritis were lower than placebo; however, changes were small and not dose-dependent.
After treatment with XELJANZ in patients with rheumatoid arthritis, rapid decreases in serum C-reactive protein (CRP) were observed and maintained throughout dosing. Changes in CRP observed with XELJANZ treatment do not reverse fully within 2 weeks after discontinuation, indicating a longer duration of pharmacodynamic activity compared to the pharmacokinetic half-life.
Similar changes in T cells, B cells, and serum CRP have been observed in patients with active psoriatic arthritis although reversibility was not assessed. Total serum immunoglobulins were not assessed in patients with active psoriatic arthritis.
12.3 Pharmacokinetics
XELJANZ/XELJANZ Oral Solution
Following oral administration of XELJANZ/XELJANZ Oral Solution, peak plasma concentrations are reached within 0.5–1 hour, elimination half-life is about 3 hours and a dose-proportional increase in systemic exposure was observed in the therapeutic dose range. Steady state concentrations are achieved in 24–48 hours with negligible accumulation after twice daily administration.
XELJANZ XR
Following oral administration of XELJANZ XR, peak plasma concentrations are reached at 4 hours and half-life is about 6 to 8 hours. Steady state concentrations are achieved within 48 hours with negligible accumulation after once daily administration.
| PK Parameters* (CV%) | XELJANZ | XELJANZ XR | ||
|---|---|---|---|---|
| Dosing Regimen | 5 mg Twice Daily | 10 mg Twice Daily | 11 mg Once Daily | 22 mg Once Daily |
| Abbreviations: AUC24 = area under the concentration-time profile from time 0 to 24 hours; Cmax = maximum plasma concentration; Cmin = minimum plasma concentration; Tmax = time to Cmax; CV = Coefficient of variation. | ||||
AUC24 (ng.hr/mL) | 263.4 (15) | 539.6 (22) | 269.0 (18) | 596.6 (19) |
Cmax (ng/mL) | 42.7 (26) | 84.7 (18) | 38.2 (15) | 83.8 (25) |
Cmin (ng/mL) | 1.41 (40) | 3.10 (54) | 1.07 (69) | 3.11 (43) |
Tmax (hours) | 1.0 | 0.8 | 4.0 | 4.0 |
Absorption
XELJANZ
The absolute oral bioavailability of XELJANZ is 74%. Coadministration of XELJANZ with a high-fat meal resulted in no changes in AUC while Cmax was reduced by 32%. In clinical trials, XELJANZ was administered without regard to meals [see Dosage and Administration (2.1)].
Distribution
After intravenous administration, the volume of distribution is 87 L. The protein binding of tofacitinib is approximately 40%. Tofacitinib binds predominantly to albumin and does not appear to bind to α1-acid glycoprotein. Tofacitinib distributes equally between red blood cells and plasma.
Metabolism and Excretion
Clearance mechanisms for tofacitinib are approximately 70% hepatic metabolism and 30% renal excretion of the parent drug. The metabolism of tofacitinib is primarily mediated by CYP3A4 with minor contribution from CYP2C19. In a human radiolabeled study, more than 65% of the total circulating radioactivity was accounted for by unchanged tofacitinib, with the remaining 35% attributed to 8 metabolites, each accounting for less than 8% of total radioactivity. The pharmacologic activity of tofacitinib is attributed to the parent molecule.
Pharmacokinetics in Patient Populations
Population pharmacokinetic analyses indicated that pharmacokinetic characteristics were similar between patients with rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis, and UC. The coefficient of variation (%) in AUC of tofacitinib were generally similar across different disease patients, ranging from 22% to 34% (Table 8).
| Pharmacokinetic Parameters* Geometric Mean (CV%) | XELJANZ 5 mg Twice Daily | XELJANZ 10 mg Twice Daily | |||
|---|---|---|---|---|---|
| Rheumatoid Arthritis | Psoriatic Arthritis | Ankylosing Spondylitis | Ulcerative Colitis | Ulcerative Colitis | |
| Abbreviations: AUC0–24,ss=area under the plasma concentration-time curve over 24 hours at steady state; CV=coefficient of variation. | |||||
| |||||
AUC0–24,ss | 504 | 419 | 381 | 423 | 807 |
Specific Populations
Covariate evaluation as part of population PK analyses in adult patient populations indicated no clinically relevant change in tofacitinib exposure, after accounting for differences in renal function (i.e., creatinine clearance) between patients, based on age, weight, gender and race (Figure 1). An approximately linear relationship between body weight and volume of distribution was observed, resulting in higher peak (Cmax) and lower trough (Cmin) concentrations in lighter patients. However, this difference is not considered to be clinically relevant.
Covariate evaluation as part of population PK analyses in pcJIA patients identified body weight significantly impacting tofacitinib exposure, which supports weight-based dosing in this population. No additional dose adjustment is needed based on age, gender, race, or disease severity in pcJIA patients.
The effect of renal and hepatic impairment and other intrinsic factors on the pharmacokinetics of tofacitinib is shown in Figure 1.
Figure 1: Impact of Intrinsic Factors on Tofacitinib Pharmacokinetics
In subjects with ESRD maintained on hemodialysis, mean AUC was approximately 40% higher compared with historical healthy subject data, consistent with approximately 30% contribution of renal clearance to the total clearance of tofacitinib. Dose adjustment is recommended in RA, PsA, AS, UC, and pcJIA patients with ESRD maintained on hemodialysis [see Dosage and Administration (2.2, 2.3, 2.4)].
Drug Interaction Studies
Potential for XELJANZ/XELJANZ XR/XELJANZ Oral Solution to Influence the PK of Other Drugs
In vitro studies indicate that tofacitinib does not significantly inhibit or induce the activity of the major human drug-metabolizing CYPs (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) at concentrations corresponding to the steady state Cmax of a 10 mg twice daily dose. These in vitro results were confirmed by a human drug interaction study showing no changes in the pharmacokinetics of midazolam, a highly sensitive CYP3A4 substrate, when coadministered with XELJANZ.
In vitro studies indicate that tofacitinib does not significantly inhibit the activity of the major human drug-metabolizing uridine 5'-diphospho-glucuronosyltransferases (UGTs) [UGT1A1, UGT1A4, UGT1A6, UGT1A9, and UGT2B7] at concentrations exceeding 250 times the steady state Cmax of a 10 mg twice daily dose.
In rheumatoid arthritis patients, the oral clearance of tofacitinib does not vary with time, indicating that tofacitinib does not normalize CYP enzyme activity in rheumatoid arthritis patients. Therefore, coadministration with XELJANZ/XELJANZ XR is not expected to result in clinically relevant increases in the metabolism of CYP substrates in rheumatoid arthritis patients.
In vitro data indicate that the potential for tofacitinib to inhibit transporters such as P-glycoprotein, organic anionic or cationic transporters at therapeutic concentrations is low.
Dosing recommendations for coadministered drugs following administration with XELJANZ/XELJANZ XR/XELJANZ Oral Solution are shown in Figure 2.
Figure 2: Impact of Tofacitinib on the Pharmacokinetics of Other Drugs
| Note: Reference group is administration of concomitant medication alone; OCT = Organic Cationic Transporter; MATE = Multidrug and Toxic Compound Extrusion. |
|
Potential for Other Drugs to Influence the Pharmacokinetics of Tofacitinib
Since tofacitinib is metabolized by CYP3A4, interaction with drugs that inhibit or induce CYP3A4 is likely. Inhibitors of CYP2C19 alone or P-glycoprotein are unlikely to substantially alter the pharmacokinetics of tofacitinib (see Figure 3).
Figure 3: Impact of Other Drugs on the Pharmacokinetics of Tofacitinib
| Note: Reference group is administration of tofacitinib alone. a [see Dosage and Administration (2.2, 2.3, 2.4), Drug Interactions (7)]. |
|
Health Professional Information
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.