SYNAREL® Adverse Reactions
(nafarelin acetate)
ADVERSE REACTIONS
In clinical trials of 155 pediatric patients, 2.6% reported symptoms suggestive of drug sensitivity, such as shortness of breath, chest pain, urticaria, rash, and pruritus.
In these 155 patients treated for an average of 41 months and as long as 80 months (6.7 years), adverse events most frequently reported (>3% of patients) consisted largely of episodes occurring during the first 6 weeks of treatment as a result of the transient stimulatory action of nafarelin upon the pituitary-gonadal axis:
- acne (10%)
- transient breast enlargement (8%)
- vaginal bleeding (8%)
- emotional lability (6%) [see Warnings]
- transient increase in pubic hair (5%)
- body odor (4%)
- seborrhea (3%)
Hot flashes, common in adult women treated for endometriosis, occurred in only 3% of treated children and were transient. Other adverse events thought to be drug-related, and occurring in >3% of patients were rhinitis (5%) and white or brownish vaginal discharge (3%). Approximately 3% of patients withdrew from clinical trials due to adverse events.
In one male patient with concomitant congenital adrenal hyperplasia, and who had discontinued treatment 8 months previously to resume puberty, adrenal rest tumors were found in the left testis. Relationship to SYNAREL is unlikely.
Regular examinations of the pituitary gland by magnetic resonance imaging (MRI) or computer assisted tomography (CT) of children during long-term nafarelin therapy as well as during the post-treatment period has occasionally revealed changes in the shape and size of the pituitary gland. These changes include asymmetry and enlargement of the pituitary gland, and a pituitary microadenoma has been suspected in a few children. The relationship of these findings to SYNAREL is not known.
Post-Marketing
Pituitary apoplexy: During post-marketing surveillance, rare cases of pituitary apoplexy (a clinical syndrome secondary to infarction of the pituitary gland) have been reported after the administration of gonadotropin-releasing hormone agonists. In a majority of these cases, a pituitary adenoma was diagnosed, with a majority of pituitary apoplexy cases occurring within 2 weeks of the first dose, and some within the first hour. In these cases, pituitary apoplexy has presented as sudden headache, vomiting, visual changes, ophthalmoplegia, altered mental status, and sometimes cardiovascular collapse. Immediate medical attention has been required.
Psychiatric adverse events: Emotional lability, such as crying, irritability, impatience, anger, and aggression has been observed with GnRH agonists [see Warnings]; Depression, including rare reports of suicidal ideation and attempt, has been reported for GnRH agonists in children treated for central precocious puberty. Many, but not all, of these patients had a history of psychiatric illness or other comorbidities with an increased risk of depression.
ADVERSE REACTIONS
Clinical Studies
In formal clinical trials of 1509 healthy adult patients, symptoms suggestive of drug sensitivity, such as shortness of breath, chest pain, urticaria, rash and pruritus occurred in 3 patients (approximately 0.2%).
As would be expected with a drug which lowers serum estradiol levels, the most frequently reported adverse reactions were those related to hypoestrogenism.
In controlled studies comparing SYNAREL (400 µg/day) and danazol (600 or 800 mg/day), adverse reactions most frequently reported and thought to be drug-related are shown in the figure below:
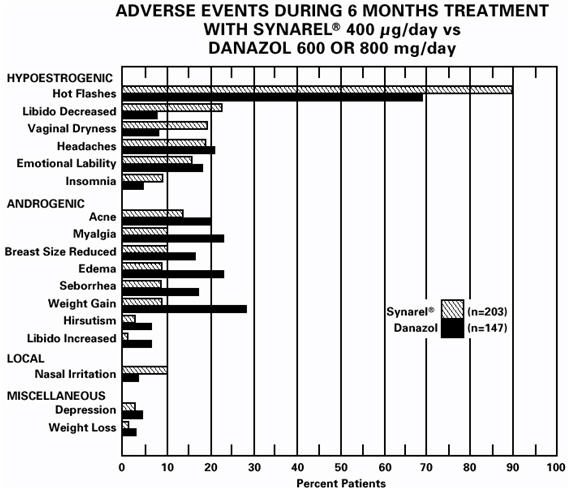
In addition, less than 1% of patients experienced paresthesia, palpitations, chloasma, maculopapular rash, eye pain, asthenia, lactation, breast engorgement, and arthralgia.
Changes in Bone Density
After six months of treatment with SYNAREL, vertebral trabecular bone density and total vertebral bone mass, measured by quantitative computed tomography (QCT), decreased by an average of 8.7% and 4.3%, respectively, compared to pretreatment levels. There was partial recovery of bone density in the post-treatment period; the average trabecular bone density and total bone mass were 4.9% and 3.3% less than the pretreatment levels, respectively. Total vertebral bone mass, measured by dual photon absorptiometry (DPA), decreased by a mean of 5.9% at the end of treatment.
After six months treatment with SYNAREL, bone mass as measured by dual x-ray bone densitometry (DEXA), decreased 3.2%. Mean total vertebral mass, re-examined by DEXA six months after completion of treatment, was 1.4% below pretreatment. There was little, if any, decrease in the mineral content in compact bone of the distal radius and second metacarpal. Use of SYNAREL for longer than the recommended six months or in the presence of other known risk factors for decreased bone mineral content may cause additional bone loss.
Changes in Laboratory Values During Treatment
Plasma enzymes
During clinical trials with SYNAREL, regular laboratory monitoring revealed that SGOT and SGPT levels were more than twice the upper limit of normal in only one patient each. There was no other clinical or laboratory evidence of abnormal liver function and levels returned to normal in both patients after treatment was stopped.
Lipids
At enrollment, 9% of the patients in the group taking SYNAREL 400 µg/day and 2% of the patients in the danazol group had total cholesterol values above 250 mg/dL. These patients also had cholesterol values above 250 mg/dL at the end of treatment.
Of those patients whose pretreatment cholesterol values were below 250 mg/dL, 6% in the group treated with SYNAREL and 18% in the danazol group, had post-treatment values above 250 mg/dL.
The mean (± SEM) pretreatment values for total cholesterol from all patients were 191.8 (4.3) mg/dL in the group treated with SYNAREL and 193.1 (4.6) mg/dL in the danazol group. At the end of treatment, the mean values for total cholesterol from all patients were 204.5 (4.8) mg/dL in the group treated with SYNAREL and 207.7 (5.1) mg/dL in the danazol group. These increases from the pretreatment values were statistically significant (p<0.05) in both groups.
Triglycerides were increased above the upper limit of 150 mg/dL in 12% of the patients who received SYNAREL and in 7% of the patients who received danazol.
At the end of treatment, no patients receiving SYNAREL had abnormally low HDL cholesterol fractions (less than 30 mg/dL) compared with 43% of patients receiving danazol. None of the patients receiving SYNAREL had abnormally high LDL cholesterol fractions (greater than 190 mg/dL) compared with 15% of those receiving danazol. There was no increase in the LDL/HDL ratio in patients receiving SYNAREL, but there was approximately a 2-fold increase in the LDL/HDL ratio in patients receiving danazol.
Other changes
In comparative studies, the following changes were seen in approximately 10% to 15% of patients. Treatment with SYNAREL was associated with elevations of plasma phosphorus and eosinophil counts, and decreases in serum calcium and WBC counts. Danazol therapy was associated with an increase of hematocrit and WBC.
Post-Marketing
Pituitary apoplexy: During post-marketing surveillance, rare cases of pituitary apoplexy (a clinical syndrome secondary to infarction of the pituitary gland) have been reported after the administration of gonadotropin-releasing hormone agonists. In a majority of these cases, a pituitary adenoma was diagnosed, with a majority of pituitary apoplexy cases occurring within 2 weeks of the first dose, and some within the first hour. In these cases, pituitary apoplexy has presented as sudden headache, vomiting, visual changes, ophthalmoplegia, altered mental status, and sometimes cardiovascular collapse. Immediate medical attention has been required.
Cardiovascular adverse events: Cases of serious venous and arterial thromboembolism have been reported, including deep vein thrombosis, pulmonary embolism, myocardial infarction, stroke, and transient ischemic attack. Although a temporal relationship was reported in some cases, most cases were confounded by risk factors or concomitant medication use. It is unknown if there is a causal association between the use of GnRH analogs and these events.
Find SYNAREL® medical information:
Find SYNAREL® medical information:
SYNAREL® Quick Finder
Health Professional Information
Adverse Reactions
ADVERSE REACTIONS
In clinical trials of 155 pediatric patients, 2.6% reported symptoms suggestive of drug sensitivity, such as shortness of breath, chest pain, urticaria, rash, and pruritus.
In these 155 patients treated for an average of 41 months and as long as 80 months (6.7 years), adverse events most frequently reported (>3% of patients) consisted largely of episodes occurring during the first 6 weeks of treatment as a result of the transient stimulatory action of nafarelin upon the pituitary-gonadal axis:
- acne (10%)
- transient breast enlargement (8%)
- vaginal bleeding (8%)
- emotional lability (6%) [see Warnings]
- transient increase in pubic hair (5%)
- body odor (4%)
- seborrhea (3%)
Hot flashes, common in adult women treated for endometriosis, occurred in only 3% of treated children and were transient. Other adverse events thought to be drug-related, and occurring in >3% of patients were rhinitis (5%) and white or brownish vaginal discharge (3%). Approximately 3% of patients withdrew from clinical trials due to adverse events.
In one male patient with concomitant congenital adrenal hyperplasia, and who had discontinued treatment 8 months previously to resume puberty, adrenal rest tumors were found in the left testis. Relationship to SYNAREL is unlikely.
Regular examinations of the pituitary gland by magnetic resonance imaging (MRI) or computer assisted tomography (CT) of children during long-term nafarelin therapy as well as during the post-treatment period has occasionally revealed changes in the shape and size of the pituitary gland. These changes include asymmetry and enlargement of the pituitary gland, and a pituitary microadenoma has been suspected in a few children. The relationship of these findings to SYNAREL is not known.
Post-Marketing
Pituitary apoplexy: During post-marketing surveillance, rare cases of pituitary apoplexy (a clinical syndrome secondary to infarction of the pituitary gland) have been reported after the administration of gonadotropin-releasing hormone agonists. In a majority of these cases, a pituitary adenoma was diagnosed, with a majority of pituitary apoplexy cases occurring within 2 weeks of the first dose, and some within the first hour. In these cases, pituitary apoplexy has presented as sudden headache, vomiting, visual changes, ophthalmoplegia, altered mental status, and sometimes cardiovascular collapse. Immediate medical attention has been required.
Psychiatric adverse events: Emotional lability, such as crying, irritability, impatience, anger, and aggression has been observed with GnRH agonists [see Warnings]; Depression, including rare reports of suicidal ideation and attempt, has been reported for GnRH agonists in children treated for central precocious puberty. Many, but not all, of these patients had a history of psychiatric illness or other comorbidities with an increased risk of depression.
ADVERSE REACTIONS
Clinical Studies
In formal clinical trials of 1509 healthy adult patients, symptoms suggestive of drug sensitivity, such as shortness of breath, chest pain, urticaria, rash and pruritus occurred in 3 patients (approximately 0.2%).
As would be expected with a drug which lowers serum estradiol levels, the most frequently reported adverse reactions were those related to hypoestrogenism.
In controlled studies comparing SYNAREL (400 µg/day) and danazol (600 or 800 mg/day), adverse reactions most frequently reported and thought to be drug-related are shown in the figure below:
In addition, less than 1% of patients experienced paresthesia, palpitations, chloasma, maculopapular rash, eye pain, asthenia, lactation, breast engorgement, and arthralgia.
Changes in Bone Density
After six months of treatment with SYNAREL, vertebral trabecular bone density and total vertebral bone mass, measured by quantitative computed tomography (QCT), decreased by an average of 8.7% and 4.3%, respectively, compared to pretreatment levels. There was partial recovery of bone density in the post-treatment period; the average trabecular bone density and total bone mass were 4.9% and 3.3% less than the pretreatment levels, respectively. Total vertebral bone mass, measured by dual photon absorptiometry (DPA), decreased by a mean of 5.9% at the end of treatment.
After six months treatment with SYNAREL, bone mass as measured by dual x-ray bone densitometry (DEXA), decreased 3.2%. Mean total vertebral mass, re-examined by DEXA six months after completion of treatment, was 1.4% below pretreatment. There was little, if any, decrease in the mineral content in compact bone of the distal radius and second metacarpal. Use of SYNAREL for longer than the recommended six months or in the presence of other known risk factors for decreased bone mineral content may cause additional bone loss.
Changes in Laboratory Values During Treatment
Plasma enzymes
During clinical trials with SYNAREL, regular laboratory monitoring revealed that SGOT and SGPT levels were more than twice the upper limit of normal in only one patient each. There was no other clinical or laboratory evidence of abnormal liver function and levels returned to normal in both patients after treatment was stopped.
Lipids
At enrollment, 9% of the patients in the group taking SYNAREL 400 µg/day and 2% of the patients in the danazol group had total cholesterol values above 250 mg/dL. These patients also had cholesterol values above 250 mg/dL at the end of treatment.
Of those patients whose pretreatment cholesterol values were below 250 mg/dL, 6% in the group treated with SYNAREL and 18% in the danazol group, had post-treatment values above 250 mg/dL.
The mean (± SEM) pretreatment values for total cholesterol from all patients were 191.8 (4.3) mg/dL in the group treated with SYNAREL and 193.1 (4.6) mg/dL in the danazol group. At the end of treatment, the mean values for total cholesterol from all patients were 204.5 (4.8) mg/dL in the group treated with SYNAREL and 207.7 (5.1) mg/dL in the danazol group. These increases from the pretreatment values were statistically significant (p<0.05) in both groups.
Triglycerides were increased above the upper limit of 150 mg/dL in 12% of the patients who received SYNAREL and in 7% of the patients who received danazol.
At the end of treatment, no patients receiving SYNAREL had abnormally low HDL cholesterol fractions (less than 30 mg/dL) compared with 43% of patients receiving danazol. None of the patients receiving SYNAREL had abnormally high LDL cholesterol fractions (greater than 190 mg/dL) compared with 15% of those receiving danazol. There was no increase in the LDL/HDL ratio in patients receiving SYNAREL, but there was approximately a 2-fold increase in the LDL/HDL ratio in patients receiving danazol.
Other changes
In comparative studies, the following changes were seen in approximately 10% to 15% of patients. Treatment with SYNAREL was associated with elevations of plasma phosphorus and eosinophil counts, and decreases in serum calcium and WBC counts. Danazol therapy was associated with an increase of hematocrit and WBC.
Post-Marketing
Pituitary apoplexy: During post-marketing surveillance, rare cases of pituitary apoplexy (a clinical syndrome secondary to infarction of the pituitary gland) have been reported after the administration of gonadotropin-releasing hormone agonists. In a majority of these cases, a pituitary adenoma was diagnosed, with a majority of pituitary apoplexy cases occurring within 2 weeks of the first dose, and some within the first hour. In these cases, pituitary apoplexy has presented as sudden headache, vomiting, visual changes, ophthalmoplegia, altered mental status, and sometimes cardiovascular collapse. Immediate medical attention has been required.
Cardiovascular adverse events: Cases of serious venous and arterial thromboembolism have been reported, including deep vein thrombosis, pulmonary embolism, myocardial infarction, stroke, and transient ischemic attack. Although a temporal relationship was reported in some cases, most cases were confounded by risk factors or concomitant medication use. It is unknown if there is a causal association between the use of GnRH analogs and these events.
Health Professional Information
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.