SOMAVERT® WITH DILUENT VIAL
(pegvisomant)
Find SOMAVERT® WITH DILUENT VIAL medical information:
Find SOMAVERT® WITH DILUENT VIAL medical information:
SOMAVERT® WITH DILUENT VIAL Quick Finder
Highlights
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use SOMAVERT safely and effectively. See full prescribing information for SOMAVERT. SOMAVERT (pegvisomant) for injection, for subcutaneous use Initial U.S. Approval: 2003 INDICATIONS AND USAGESOMAVERT is a growth hormone receptor antagonist indicated for the treatment of acromegaly in patients who have had an inadequate response to surgery or radiation therapy, or for whom these therapies are not appropriate. The goal of treatment is to normalize serum insulin-like growth factor-1 (IGF-1) levels. (1) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSFor injection: 10 mg, 15 mg, 20 mg, 25 mg or 30 mg lyophilized powder in a single-dose vial for reconstitution supplied with a single-dose vial containing diluent (Sterile Water for Injection, USP) (3) CONTRAINDICATIONSNone (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common reported adverse reactions (>6%) are infection, pain, nausea, diarrhea, abnormal liver tests, flu syndrome, injection site reaction (6) To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONSFemales and Males of Reproductive Potential: Advise premenopausal females of the potential for an unintended pregnancy (8.3) See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 7/2023 |
Indications and Usage
Dosage and Administration
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
The recommended loading dose of SOMAVERT is 40 mg given subcutaneously, under healthcare provider supervision. Provide proper training in subcutaneous injection technique to patients or their caregivers so they can receive once daily subcutaneous injections. On the next day following the loading dose, instruct patients or their caregivers to begin daily subcutaneous injections of 10 mg of SOMAVERT.
Titrate the dosage to normalize serum IGF-1 concentrations (serum IGF-1 concentrations should be measured every four to six weeks). The dosage should not be based on growth hormone (GH) concentrations or signs and symptoms of acromegaly. It is unknown whether patients who remain symptomatic while achieving normalized IGF-1 concentrations would benefit from increased SOMAVERT dosage.
- •
- Increase the dosage by 5 mg increments every 4–6 weeks if IGF-1 concentrations are elevated.
- •
- Decrease the dosage by 5 mg decrements every 4–6 weeks if IGF-1 concentrations are below the normal range.
- •
- IGF-1 levels should also be monitored when a SOMAVERT dose given in multiple injections is converted to a single daily injection [see Clinical Pharmacology (12)].
The recommended dosage range is between 10 mg to 30 mg given subcutaneously once daily and the maximum daily dosage is 30 mg given subcutaneously once daily.
2.2 Assess Liver Tests Prior to Initiation of SOMAVERT
Prior to the start of SOMAVERT, patients should have an assessment of baseline levels of liver tests [serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), serum total bilirubin (TBIL), and alkaline phosphatase (ALP)]. For recommendations regarding initiation of SOMAVERT based on baseline liver tests and recommendations for monitoring of liver tests while on SOMAVERT, refer to Table 1 in Warning and Precautions (5.2).
2.3 Loading Dose Injection Procedure
The following instructions are for the healthcare provider to reconstitute and prepare the 40 mg loading dose. The healthcare provider will need to reconstitute 1 vial of lyophilized powder of SOMAVERT containing 20 mg of pegvisomant with supplied diluent (two vials of lyophilized powder and two vials of diluent will be needed for the 40 mg loading dose). The healthcare provider will also need to inject the reconstituted SOMAVERT solution twice into the patient's upper arm, upper thigh, abdomen, or buttocks (each injection in a different area).
- a)
- Before administering the loading dose, remove the first package (1 vial of lyophilized powder of SOMAVERT containing 20 mg of pegvisomant and 1 vial containing the diluent) from the refrigerator about 10 minutes prior to the planned injection time.
- b)
- Withdraw 1 mL of the supplied diluent (Sterile Water for Injection) and inject slowly onto the sides of the vial containing lyophilized powder of SOMAVERT. Do not inject the diluent directly on the powder.
- c)
- Do not invert the vial or shake the solution as this may cause denaturation of the pegvisomant protein. Slowly swirl the solution to ensure that all of the lyophilized powder has gone into solution. If foaming of the reconstituted SOMAVERT solution is seen, the solution is likely damaged and therefore inappropriate to inject.
- d)
- Visually inspect the reconstituted SOMAVERT solution for particulate matter and discoloration prior to administration. The reconstituted solution should be clear. If the solution is cloudy, do not use it. Once reconstituted, the solution will contain 20 mg of pegvisomant in 1 mL of solution.
- e)
- Withdraw the 1 mL reconstituted SOMAVERT solution. The solution must be administered within 6 hours of reconstitution.
- f)
- Inject the first reconstituted SOMAVERT solution (20 mg/mL) subcutaneously into the patient's upper arm, upper thigh, abdomen, or buttocks using a 90-degree angle.
- g)
- Repeat steps (a) to (e) to reconstitute the second SOMAVERT dose of 20mg.
- h)
- Finally, inject the second reconstituted SOMAVERT solution (20 mg/mL) subcutaneously into the patient's upper arm, upper thigh, abdomen, or buttocks using a 90-degree angle (different area than the first injection).
2.4 Maintenance Dose Injection Procedure
For patient or caregiver instructions for reconstitution and administration of daily doses (10 mg to 30 mg), see the Patient's Instructions for Use.
- a)
- Before administering the dose, remove one package (1 vial of lyophilized powder of SOMAVERT containing 10 mg, 15 mg, 20 mg, 25 mg or 30 mg of pegvisomant and 1 vial containing the diluent) from the refrigerator about 10 minutes prior to the planned injection time.
- b)
- Withdraw 1 mL of the supplied 5 mL diluent (Sterile Water for Injection) and inject slowly onto the sides of the vial containing lyophilized powder of SOMAVERT. Do not inject the diluent directly on the powder.
- c)
- Do not invert the vial or shake the solution as this may cause denaturation of the pegvisomant protein. Slowly swirl the solution to ensure that all of the lyophilized powder has gone into solution. If foaming of the reconstituted SOMAVERT solution is seen, the solution is likely damaged and therefore inappropriate to inject.
- d)
- Visually inspect the reconstituted SOMAVERT solution for particulate matter and discoloration prior to administration. The reconstituted solution should be clear. If the solution is cloudy, do not use it. Once reconstituted, the solution will contain 10 mg, 15 mg, 20 mg, 25 mg or 30 mg of pegvisomant in 1 mL of solution.
- e)
- Withdraw the 1 mL reconstituted SOMAVERT solution. The solution must be administered within 6 hours of reconstitution.
- f)
- Inject the reconstituted SOMAVERT solution subcutaneously into the upper arm, upper thigh, abdomen, or buttocks using a 90-degree angle.
Dosage Forms and Strengths
Contraindications
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Hypoglycemia Associated With GH Lowering in Patients With Diabetes Mellitus
GH opposes the effects of insulin on carbohydrate metabolism by decreasing insulin sensitivity; thus, glucose tolerance may improve in some patients treated with SOMAVERT. Patients should be carefully monitored and doses of anti-diabetic drugs reduced as necessary to avoid hypoglycemia in patients with diabetes mellitus.
5.2 Liver Toxicity
Baseline serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), serum total bilirubin (TBIL), and alkaline phosphatase (ALP) levels should be obtained prior to initiating therapy with SOMAVERT. Table 1 lists recommendations regarding initiation of treatment with SOMAVERT, based on the results of these liver tests (LTs).
Asymptomatic, transient elevations in transaminases up to 15 times ULN have been observed in <2% of subjects among two open-label trials (with a total of 147 patients). These reports were not associated with an increase in bilirubin. Transaminase elevations normalized with time, most often after suspending treatment. Postmarketing reports have identified elevations in serum hepatic transaminases up to greater than 20 times ULN associated with elevation in total bilirubin greater than 2 times ULN. In many of these cases, discontinuation of SOMAVERT therapy resulted in improvement or resolution of hepatic laboratory abnormalities.
SOMAVERT should be used in accordance with the information presented in Table 2 with respect to liver test abnormalities while on SOMAVERT treatment.
| Baseline LT Levels | Recommendations |
|---|---|
Normal |
|
Elevated, but less than or equal to 3 times ULN | May treat with SOMAVERT; however, monitor LTs monthly for at least one year after initiation of therapy and then bi-annually for the next year. |
Greater than 3 times ULN |
|
If a patient develops LT elevations, or any other signs or symptoms of liver dysfunction while receiving SOMAVERT, the following patient management is recommended (Table 2).
| LT Levels and Clinical Signs/Symptoms | Recommendations |
|---|---|
Greater than or equal to 3 but less than 5 times ULN (without signs/symptoms of hepatitis or other liver injury, or increase in serum TBIL) |
|
At least 5 times ULN, or transaminase elevations at least 3 times ULN associated with any increase in serum TBIL (with or without signs/symptoms of hepatitis or other liver injury) |
|
Signs or symptoms suggestive of hepatitis or other liver injury (e.g., jaundice, bilirubinuria, fatigue, nausea, vomiting, right upper quadrant pain, ascites, unexplained edema, easy bruisability) |
|
5.3 Cross-Reactivity With GH Assays
SOMAVERT has significant structural similarity to growth hormone (GH) which causes it to cross-react in commercially available GH assays. Since serum concentrations of therapeutically effective doses of SOMAVERT are generally 100 to 1000 times higher than the actual serum GH concentrations seen in patients with acromegaly, measurements of serum GH concentrations will appear falsely elevated.
5.4 Lipohypertrophy
There have been cases of lipohypertrophy in patients treated with SOMAVERT. In a double-blind, 12-week, placebo-controlled study, there was one case (1.3%) of injection site lipohypertrophy reported in a subject receiving 10 mg/day. The subject recovered while on treatment. Among two open-label trials (with a total of 147 patients), there were two subjects, both receiving 10 mg/day, who developed lipohypertrophy. One case recovered while on treatment, and one case resulted in a discontinuation of treatment. Injection sites should be rotated daily to help prevent lipohypertrophy (different area than the last injection).
5.5 Systemic Hypersensitivity
In patients with systemic hypersensitivity reactions, caution and close monitoring should be exercised when re-initiating SOMAVERT therapy [see Adverse Reactions (6.2)].
Adverse Reactions
6 ADVERSE REACTIONS
Clinically significant adverse reactions that appear in other section of the labeling include:
- •
- Hypoglycemia Associated with GH Lowering in Patients with Diabetes Mellitus [see Warnings and Precautions (5.1)]
- •
- Liver Toxicity [see Warnings and Precautions (5.2)]
- •
- Cross-Reactivity with GH Assays [see Warnings and Precautions (5.3)]
- •
- Lipohypertrophy [see Warnings and Precautions (5.4)]
- •
- Systemic Hypersensitivity [see Warnings and Precautions (5.5)]
Elevations of serum concentrations of ALT and AST greater than ten times the ULN were reported in two patients (0.8%) exposed to SOMAVERT in pre-approval clinical studies. One patient was rechallenged with SOMAVERT, and the recurrence of elevated transaminase levels suggested a probable causal relationship between administration of the drug and the elevation in liver enzymes. A liver biopsy performed on the second patient was consistent with chronic hepatitis of unknown etiology. In both patients, the transaminase elevations normalized after discontinuation of the drug.
Elevations in ALT and AST levels were not associated with increased levels of TBIL and ALP, with the exception of two patients with minimal associated increases in ALP levels (i.e., less than 3 times ULN). The transaminase elevations did not appear to be related to the dose of SOMAVERT administered, generally occurred within 4 to 12 weeks of initiation of therapy, and were not associated with any identifiable biochemical, phenotypic, or genetic predictors.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In a 12-week randomized, placebo-controlled, double-blind, fixed-dose study of SOMAVERT in subjects with acromegaly, 32 subjects received placebo and 80 subjects received SOMAVERT once daily [see Clinical Studies (14)]. A total of 108 subjects (30 placebo, 78 SOMAVERT) completed 12 weeks of study treatment.
Overall, eight patients with acromegaly (5.3%) withdrew from pre-marketing clinical studies because of adverse events, including two patients with marked transaminase elevations, one patient with lipohypertrophy at the injection sites, and one patient with substantial weight gain. Most adverse events did not appear to be dose-dependent. Table 3 shows the incidence of adverse events that were reported in at least two patients treated with SOMAVERT and at frequencies greater than placebo during the 12-week, placebo-controlled study.
| Placebo n=32 | SOMAVERT | |||
|---|---|---|---|---|
| 10 mg/day n=26 | 15 mg/day n=26 | 20 mg/day N=28 | ||
| ||||
Infection† | 2 (6%) | 6 (23%) | 0 | 0 |
Pain | 2 (6%) | 2 (8%) | 1 (4%) | 4 (14%) |
Nausea | 1 (3%) | 0 | 2 (8%) | 4 (14%) |
Diarrhea | 1 (3%) | 1 (4%) | 0 | 4 (14%) |
Abnormal liver function tests | 1 (3%) | 3 (12%) | 1 (4%) | 1 (4%) |
Flu syndrome | 0 | 1 (4%) | 3 (12%) | 2 (7%) |
Injection site reaction | 0 | 2 (8%) | 1 (4%) | 3 (11%) |
Dizziness | 2 (6%) | 2 (8%) | 1 (4%) | 1 (4%) |
Accidental injury | 1 (3%) | 2 (8%) | 1 (4%) | 0 |
Back pain | 1 (3%) | 2 (8%) | 0 | 1 (4%) |
Sinusitis | 1 (3%) | 2 (8%) | 0 | 1 (4%) |
Chest pain | 0 | 1 (4%) | 2 (8%) | 0 |
Peripheral edema | 0 | 2 (8%) | 0 | 1 (4%) |
Hypertension | 0 | 0 | 2 (8%) | 0 |
Paresthesia | 2 (6%) | 0 | 0 | 2 (7%) |
6.2 Postmarketing Experience
Adverse Reactions from Postmarketing Spontaneous Reports
The following adverse reactions have been identified during post-approval use of SOMAVERT. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Systemic hypersensitivity reactions including anaphylactic reactions, laryngospasm, angioedema, generalized skin reactions (rash, erythema, pruritus, urticaria) have been reported in post-marketing use. Some patients required hospitalization. Symptoms did not re-occur in all patients after re-challenge [see Warnings and Precautions (5.5)].
Adverse Reactions from an Observational Study
ACROSTUDY was an international observational registry that captured long term safety data in 2221 patients with acromegaly treated with SOMAVERT for a mean treatment duration of 8.5 years. Patients could also receive other therapy for acromegaly during the registry period. Treatment dose and schedule were at the discretion of each treating healthcare provider. Although safety monitoring as per the recommended schedule was mandatory, not all assessments were performed at all time points for every patient. Because of this, comparison of rates of adverse events to those in the original clinical trial is not appropriate. Of the 1327 patients who had a normal AST and ALT at baseline, 20 (1.5%) patients had elevated tests >3-5 times ULN, and 22 (1.7%) patients had elevated tests >5 times ULN. Lipohypertrophy was reported in 35 (1.6%) patients. Of the 1795 patients who had a MRI reported at baseline and at least once during follow up in the study, MRI results showed that 128 (7.1%) were reported to have an increase, 310 (17.3%) were reported to have a decrease, 81 (4.5%) had both increase and decrease, and 1276 (71.1%) had no change.
Drug Interactions
7 DRUG INTERACTIONS
7.1 Insulin and/or Oral Hypoglycemic Agents
After initiation of SOMAVERT, patients with acromegaly and diabetes mellitus treated with insulin and/or oral hypoglycemic agents may require dose reductions of insulin and/or oral hypoglycemic agents [see Warnings and Precautions (5.1)].
Use in Specific Populations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Postmarketing reports of SOMAVERT use in pregnant women are insufficient to establish a drug-associated risk for major birth defects, miscarriage or adverse maternal or fetal outcomes. Acromegaly may improve during pregnancy (see Clinical Considerations). In animal reproduction studies, fetotoxicity was observed at a dose that was 6 times the maximum recommended human dose based on body surface area following subcutaneous administration of pegvisomant during organogenesis or during the preimplantation period (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Disease-associated maternal and/or embryofetal risk
Published data from case reports, case series, and a small interventional study in pregnant women with acromegaly have demonstrated that acromegaly may improve or stabilize without treatment during pregnancy, particularly if acromegaly is treated before pregnancy. In rare cases, acromegaly may worsen during pregnancy. Since IGF-1 levels may change physiologically during pregnancy and interpreting IGF-1 and growth hormone levels in pregnant women with acromegaly may be unreliable, clinical monitoring is recommended.
Animal Data
The effects of pegvisomant on early embryonic development and embryo-fetal development were evaluated in two separate studies, which were conducted in pregnant rabbits with pegvisomant at subcutaneous doses of 1, 3, and 10 mg/kg/day. There was no evidence of teratogenic effects associated with pegvisomant administration during organogenesis. At the 10 mg/kg/day dose (6 times the maximum human therapeutic dose based on body surface area), a reproducible, slight increase in post-implantation loss was observed in both studies.
8.2 Lactation
Risk Summary
Limited information from a case report in published literature reported that the level of pegvisomant in human milk was below the level of detection. There is no information available on the effects of the drug on the breastfed infant or the effects of the drug on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for SOMAVERT and any potential adverse effects on the breastfed child from SOMAVERT or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Discuss the potential for unintended pregnancy with premenopausal women as the therapeutic benefits of a reduction in growth hormone (GH) levels and normalization of insulin-like growth factor 1 (IGF-1) concentration in acromegalic females treated with pegvisomant may lead to improved fertility.
8.4 Pediatric Use
The safety and effectiveness of SOMAVERT in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of SOMAVERT did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Overdosage
10 OVERDOSAGE
In one reported incident of acute overdose with SOMAVERT during pre-marketing clinical studies, a patient self-administered 80 mg/day (2.7 times the maximum recommended maintenance dosage) for seven days. The patient experienced a slight increase in fatigue, had no other complaints, and demonstrated no significant clinical laboratory abnormalities.
In cases of overdose, administration of SOMAVERT should be discontinued and not resumed until IGF-1 levels return to within or above the normal range.
Description
11 DESCRIPTION
Pegvisomant is an analog of human growth hormone (GH) of recombinant DNA origin that acts as a GH receptor antagonist.
It contains 191 amino acid residues. The molecular weight of pegvisomant is 22 kDa. The molecular weight of the PEG portion of pegvisomant is approximately 5 kDa. The predominant molecular weights of pegvisomant are thus approximately 42, 47, and 52 kDa. The schematic shows the amino acid sequence of the pegvisomant protein (PEG polymers are shown attached to the 5 most probable attachment sites). Pegvisomant is synthesized by a specific strain of Escherichia coli bacteria that has been genetically modified by the addition of a plasmid that carries a gene for GH receptor antagonist.
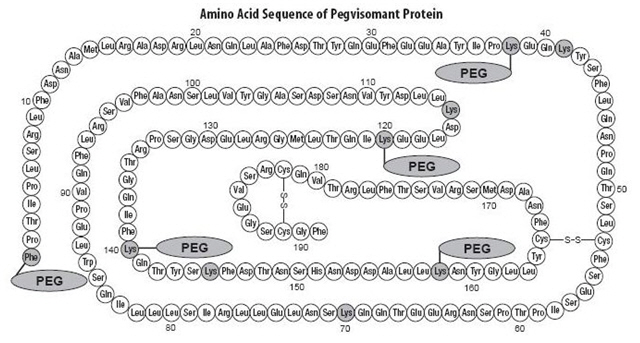
Stippled residues indicate PEG attachment sites (Phe1, Lys38, Lys41, Lys70, Lys115, Lys120, Lys140, Lys145, Lys158)
Shown below are the amino acid substitutions in pegvisomant, relative to human GH.
| hGH | Pegvisomant |
|---|---|
His18 | Asp18 |
Ala21 | Asn21 |
Gly120 | Lys120 |
Arg167 | Asn167 |
Lys168 | Ala168 |
Asp171 | Ser171 |
Lys172 | Arg172 |
Glu174 | Ser174 |
Ile179 | Thr179 |
SOMAVERT (pegvisomant) for injection is a sterile, white lyophilized powder intended for subcutaneous injection after reconstitution. SOMAVERT is supplied in packages that include a separate glass vial containing Sterile Water for Injection, USP, that is a sterile, nonpyrogenic preparation of water for injection that contains no bacteriostat, antimicrobial agent, or added buffer, to be used as a diluent.
SOMAVERT is available in single-dose sterile vials containing 10 mg, 15 mg, 20 mg, 25 mg or 30 mg of pegvisomant. SOMAVERT 10 mg, 15 mg, and 20 mg vials also contain glycine (1.36 mg), mannitol (36 mg), sodium dihydrogen phosphate monohydrate (0.36 mg), and sodium phosphate dibasic anhydrous (1.04 mg). After reconstitution with 1 mL of Water for Injection, USP, the resulting concentration is 10 mg/mL, 15 mg/mL and 20 mg/mL, respectively, with a pH of 7.1 – 7.7.
SOMAVERT 25 mg vials also contain glycine (1.7 mg), mannitol (45 mg), sodium dihydrogen phosphate monohydrate (0.45 mg), and sodium phosphate dibasic anhydrous (1.3 mg). After reconstitution with 1 mL of Water for Injection, USP, the resulting concentration is 25 mg/mL with a pH of 7.1 – 7.7.
SOMAVERT 30 mg vials also contain glycine (2.04 mg), mannitol (54 mg), sodium dihydrogen phosphate monohydrate (0.54 mg), and sodium phosphate dibasic anhydrous (1.56 mg). After reconstitution with 1 mL of Water for Injection, USP, the resulting concentration is 30 mg/mL with a pH of 7.1 – 7.7.
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pegvisomant selectively binds to growth hormone (GH) receptors on cell surfaces, where it blocks the binding of endogenous GH, and thus interferes with GH signal transduction.
Inhibition of GH action results in decreased serum concentrations of IGF-1, as well as other GH-responsive serum proteins such as free IGF-1, the acid-labile subunit of IGF-1 (ALS), and insulin-like growth factor binding protein-3 (IGFBP-3).
12.2 Pharmacodynamics
Pegvisomant binds selectively to the GH receptor, and does not cross-react with 19 other cytokine receptors tested, including prolactin. Pegvisomant leads to decreased serum concentrations of IGF-1, free IGF-1, ALS, and IGFBP-3 [see Clinical Studies (14, Figure 1)].
12.3 Pharmacokinetics
Absorption: Following subcutaneous administration, peak serum pegvisomant concentrations are not generally attained until 33 to 77 hours after administration. The mean extent of absorption of a 20-mg subcutaneous dose was 57%, relative to a 10-mg intravenous dose.
Distribution: The mean apparent volume of distribution of pegvisomant is 7 L (12% coefficient of variation), suggesting that pegvisomant does not distribute extensively into tissues. After a single subcutaneous administration, exposure (Cmax, AUC) to pegvisomant increases disproportionately with increasing dose. Mean ± SEM serum pegvisomant concentrations after 12 weeks of therapy with daily doses of 10, 15, and 20 mg were 6600 ± 1330; 16,000 ± 2200; and 27,000 ± 3100 ng/mL, respectively.
The relative bioavailability of 1 × 30 mg pegvisomant was compared to 2 × 15 mg pegvisomant in a single dose study. The AUCinf and Cmax of pegvisomant when administered as one injection of 30 mg strength was approximately 6% and 4% greater, respectively, as compared to when administered as two injections of 15 mg strengths.
Metabolism and Elimination: The pegvisomant molecule contains covalently bound polyethylene glycol polymers in order to reduce the clearance rate. Clearance of pegvisomant following multiple doses is lower than seen following a single dose. The mean total body systemic clearance of pegvisomant following multiple doses is estimated to range between 36 to 28 mL/h for subcutaneous doses ranging from 10 to 20 mg/day, respectively. Clearance of pegvisomant was found to increase with body weight. Pegvisomant is eliminated from serum with a mean half-life estimates ranging from 60 to 138 hours following either single or multiple doses. Less than 1% of administered drug is recovered in the urine over 96 hours. The elimination route of pegvisomant has not been studied in humans.
Drug Interaction Studies
In clinical studies, patients on opioids often needed higher serum pegvisomant concentrations to achieve appropriate IGF-1 suppression compared with patients not receiving opioids. The mechanism of this interaction is not known [see Drug Interactions (7.2)].
Specific Populations
No pharmacokinetic studies have been conducted in patients with renal impairment, patients with hepatic impairment, geriatric patients, or pediatric patients and the effects of race on the pharmacokinetics of pegvisomant has not been studied. No gender effect on the pharmacokinetics of pegvisomant was found in a population pharmacokinetic analysis.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of SOMAVERT or other growth hormone analogs.
In pre-marketing clinical studies, approximately 17% of the SOMAVERT-treated patients developed low titer, non‑neutralizing anti-GH antibodies. Although the presence of these antibodies did not appear to impact the efficacy of SOMAVERT, the long term clinical significance of these antibodies is not known. No assay for anti-pegvisomant antibodies is commercially available for patients receiving SOMAVERT.
The data above reflect the percentage of patients whose test results were considered positive for antibodies to SOMAVERT. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to SOMAVERT with the incidence of antibodies to other products may be misleading.
Nonclinical Toxicology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Pegvisomant was administered subcutaneously to rats daily for 2 years at doses of 2, 8, and 20 mg/kg (about 2, 9, and 22-fold a single 30 mg dose in humans on an AUC basis). Long term treatment with pegvisomant at 8 and 20 mg/kg caused an increase in malignant fibrous histiocytoma at injection sites in males. Injection site tumors were not seen in female rats at the same doses. The increased incidence of injection site tumors was most probably caused by irritation and the high sensitivity of the rat to repeated subcutaneous injections.
Clinical Studies
14 CLINICAL STUDIES
A total of one hundred twelve patients (63 men and 49 women) with acromegaly participated in a 12-week, randomized, double-blind, multi-center study comparing placebo and SOMAVERT. The mean ±SD age was 48±14 years, and the mean duration of acromegaly was 8±8 years. Ninety three had undergone previous pituitary surgery, of which 57 had also been treated with conventional radiation therapy. Six patients had undergone irradiation without surgery, nine had received only drug therapy, and four had received no previous therapy. At study start, the mean ± SD time since the subjects' last surgery and/or irradiation therapy, respectively, was 6.8 ± 0.93 years (n=63) and 5.6 ± 0.57 years (n=93).
Subjects were qualified for enrollment if their serum IGF-1, drawn after the required drug washout period, was ≥1.3 times the upper limit of the age-adjusted normal range. They were randomly assigned at the baseline visit to one of four treatment groups: placebo (n=32), 10 mg/day (n=26), 15 mg/day (n= 26), or 20 mg/day (n=28) of SOMAVERT subcutaneously IGF-1. The primary efficacy endpoint was IGF-1 percent change in IGF-1 concentrations from baseline to week 12. The three groups that received SOMAVERT showed statistically significant (p<0.01) reductions in serum levels of IGF-1 compared with the placebo group (Table 4).
| Placebo n=31 | SOMAVERT | |||
|---|---|---|---|---|
| 10 mg/day n=26 | 15 mg/day n=26 | 20 mg/day n=28 | ||
| ||||
Mean baseline IGF-1 (ng/mL) (SD) | 670 (288) | 627 (251) | 649 (293) | 732 (205) |
Mean percent change from baseline in IGF-1 (SD) | -4.0 (17) | -27 (28) | -48 (26) | -63 (21) |
SOMAVERT minus Placebo | -23* | -44* | -59* | |
There were also reductions in serum levels of free IGF-1, IGFBP-3, and ALS compared with placebo at all post-baseline visits (Figure 1).
Figure 1. Effects of SOMAVERT on Serum Markers (Mean ± Standard Error) |
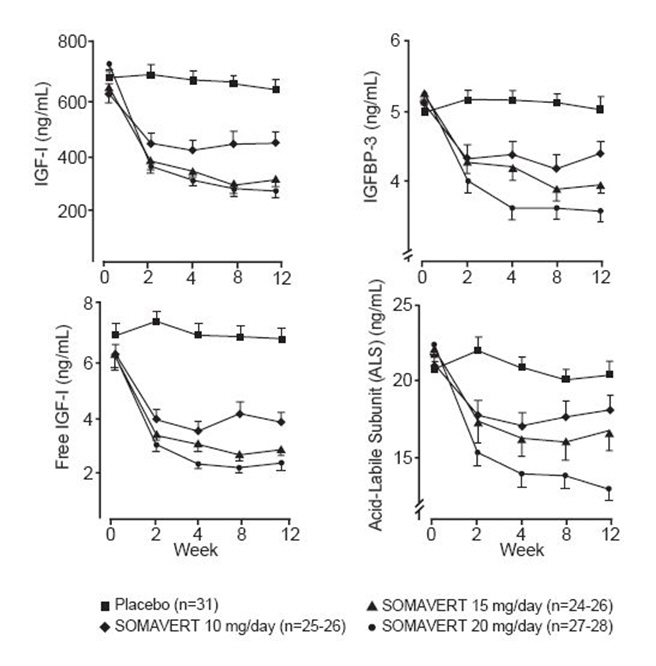 |
After 12 weeks of treatment, the following percentages of patients had normalized IGF-1 (Figure 2):
Figure 2. Percent of Patients Whose IGF-1 Levels Normalized at Week 12 |
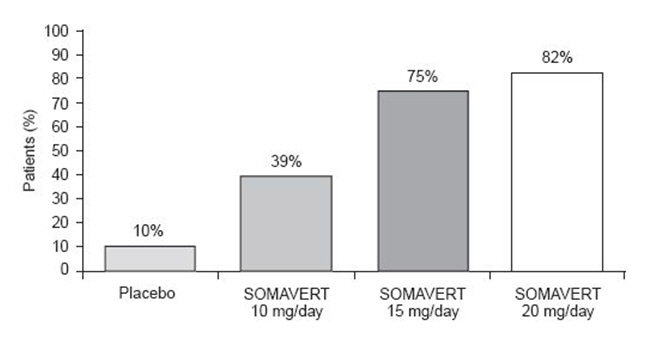 |
Table 5 shows the effect of treatment with SOMAVERT on ring size (standard jeweler's sizes converted to a numeric score ranging from 1 to 63), and on signs and symptoms of acromegaly. Each individual score for a sign or symptom of acromegaly (for soft-tissue swelling, arthralgia, headache, perspiration and fatigue) was based on a nine-point ordinal rating scale (0 = absent and 8 = severe and incapacitating), and the total score for signs or symptoms of acromegaly was derived from the sum of the individual scores. Mean baseline scores were as follows: ring size = 47.1; total signs and symptoms = 15.2; soft tissue swelling = 2.5; arthralgia = 3.2; headache = 2.4; perspiration = 3.3; and fatigue = 3.7.
| Placebo n=30 | SOMAVERT | |||
|---|---|---|---|---|
| 10 mg/day n=26 | 15 mg/day n=24–25 | 20 mg/day n=26–27 | ||
Ring size | -0.1 (2.3) | -0.8 (1.6) | -1.9 (2.0) | -2.5 (3.3) |
Total score for signs and symptoms of acromegaly | 1.3 (6.0) | -2.5 (4.3) | -4.4 (5.9) | -4.7 (4.7) |
Soft-tissue swelling | 0.3 (2.3) | -0.7 (1.6) | -1.2 (2.3) | -1.3 (1.3) |
Arthralgia | 0.1 (1.8) | -0.3 (1.8) | -0.5 (2.5) | -0.4 (2.1) |
Headache | 0.1 (1.7) | -0.4 (1.6) | -0.3 (1.4) | -0.3 (2.0) |
Perspiration | 0.1 (1.7) | -0.6 (1.6) | -1.1 (1.3) | -1.7 (1.6) |
Fatigue | 0.7 (1.5) | -0.5 (1.4) | -1.3 (1.7) | -1.0 (1.6) |
Serum growth hormone (GH) concentrations, as measured by research assays using antibodies that do not cross-react with pegvisomant, rose within two weeks of beginning treatment with SOMAVERT. The largest increase in GH concentration was seen in patients treated with doses of SOMAVERT 20 mg/day. This effect is presumably the result of diminished inhibition of GH secretion as IGF-1 levels fall. As shown in Figure 3, when patients with acromegaly were given a loading dose of SOMAVERT followed by a fixed daily dose, the rise in GH was inversely proportional to the fall in IGF-1 and generally stabilized by week 2. Serum GH concentrations remained stable in patients treated with SOMAVERT for the average of 43 weeks (range, 0–82 weeks).
Figure 3. Percent Change in Serum GH and IGF-1 Concentrations |
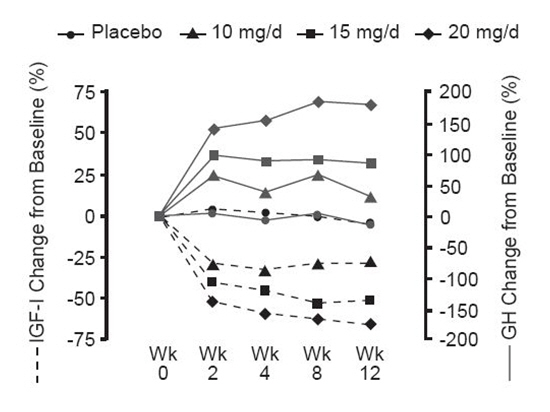 |
In the open-label extension to the clinical study, 109 subjects (including 6 new patients) with mean treatment exposure of 42.6 weeks (range 1 day – 82 weeks), 93 (85.3%) subjects had an adverse event, 16 (14.7%) had an SAE, and 4 (3.7%) discontinued due to an AE (headaches, elevated liver function tests, pancreatic cancer, and weight gain). A total of 100 (92.6%) of the 108 subjects with available IGF-1 data had a normal IGF-1 concentration at any visit during the study.
How Supplied/Storage and Handling
16 HOW SUPPLIED/STORAGE AND HANDLING
SOMAVERT (pegvisomant) for injection is a white lyophilized powder supplied in the following strengths and package configurations:
SOMAVERT (pegvisomant) for injection
| Package Configuration | NDC |
|---|---|
10 mg dose vial | NDC 0009-5176-02 |
15 mg dose vial | NDC 0009-5178-02 |
20 mg dose vial | NDC 0009-5180-02 |
25 mg dose vial | NDC 0009-5199-01 |
30 mg dose vial | NDC 0009-5200-01 |
Each package of SOMAVERT also includes a single-dose vial containing 5 mL of Sterile Water for Injection, USP.
Patient Counseling Information
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Inform patients (and/or their caregivers) of the following information to aid in the safe and effective use of SOMAVERT:
- •
- Not to use SOMAVERT if they are allergic to SOMAVERT or anything in it.
- •
- They will need blood testing to check IGF-1 levels and liver tests before and during treatment with SOMAVERT and that the dose of SOMAVERT may be changed based on the results of these tests.
- •
- SOMAVERT has not been studied in pregnant women and instruct them to notify their healthcare provider as soon as they are aware that they are pregnant.
- •
- It is not known whether SOMAVERT is excreted in human milk and instruct them to notify their healthcare provider if they plan to do so.
- •
- Pregnancy: Inform female patients that treatment with SOMAVERT may result in unintended pregnancy [see Females and Males of Reproductive Potential (8.3)].
Advise patients (and/or their caregivers) of the following adverse reactions:
- •
- The most common reported adverse reactions are injection site reaction, elevations of liver tests, pain, nausea, and diarrhea.
- •
- If they have liver test elevations they may need to have more frequent liver tests and/or discontinue SOMAVERT. Instruct patients to immediately discontinue therapy and contact their physician if they become jaundiced.
- •
- GH-secreting tumors may enlarge in people with acromegaly and that these tumors need to be watched carefully and monitored by MRI imaging.
- •
- Thickening under the skin may occur at the injection site that could lead to lumps and that switching sites may prevent or lessen this.
- •
- If they have diabetes mellitus, they may require careful monitoring and dose reductions of insulin and/or oral hypoglycemic agents while on SOMAVERT.
- •
- If they take opioids, they may need higher SOMAVERT doses to achieve appropriate IGF-1 suppression.
Advise patients that SOMAVERT is supplied as lyophilized powder in different strengths of 10 mg, 15 mg, 20 mg, 25 mg, and 30 mg in a sterile glass vial within a package also containing a single-dose flip top vial of sterile water (diluent) for injection. Advise patients that the stoppers on both vials are not made with natural rubber latex. Advise patients to follow the directions for reconstitution provided with each package including shaking may cause denaturation (destruction) of the active ingredient (therefore do not shake).
Advise patients that the package of SOMAVERT should be stored in a refrigerator 2°C to 8°C (36°F to 46°F) prior to use. It should NOT BE FROZEN.
Medication Guide

Instructions For Use
INSTRUCTIONS FOR USE
SOMAVERT (SOM-ah-vert)
(pegvisomant)
for injection, for subcutaneous use
Read this Instructions for Use before you start using SOMAVERT and each time you get a refill. There may be new information. This leaflet does not take the place of talking to your healthcare provider about your medical condition or your treatment. Your healthcare provider should show you or a caregiver how to inject SOMAVERT the right way before you inject it for the first time.
Important Information:
- •
- SOMAVERT is for use under the skin only (subcutaneous).
- •
- Do not share your SOMAVERT syringes or needles with anyone else. You may give an infection to them or get an infection from them.
- •
- Do not use SOMAVERT after the expiration date (EXP) on the label.
- •
- Inject SOMAVERT within 6 hours of mixing it. If you wait more than 6 hours, you must throw away the medicine without injecting it.
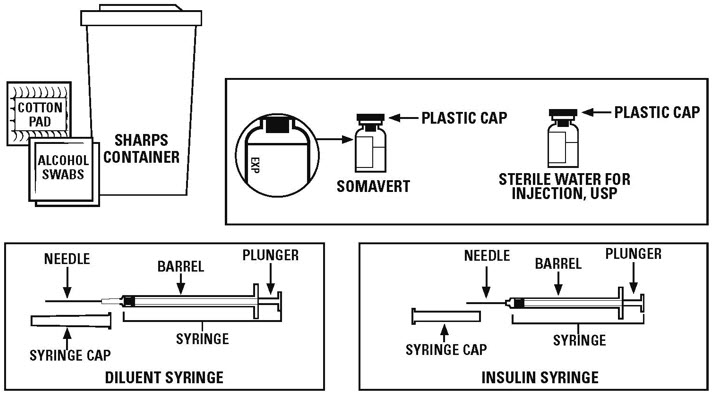
Supplies you need to give each injection of SOMAVERT. See Figure A.
- •
- 1 package of SOMAVERT that contains:
- o
- 1 vial of powdered SOMAVERT medicine (powder vial)
- o
- 1 vial of liquid (diluent) labeled "Sterile Water for Injection, USP" to mix the powdered medicine
- The vials in the package of SOMAVERT have stoppers that are not made with natural rubber latex.
- The SOMAVERT package does not come with syringes and needles.
- •
- a 1 mL syringe with a 21 gauge to 27 gauge needle that is at least 1 inch long. This is the syringe and needle needed to mix the medicine (diluent syringe).
- •
- a 1 mL insulin syringe with attached needle. This is the syringe needed for your injection.
- •
- 2 alcohol swabs
- •
- 1 small clean, dry cotton pad
- •
- 1 sharps disposal container for throwing away used needles and syringes. See "Disposing of used needles and syringes" at the end of these instructions.
- •
- a clean, flat surface to work on, like a table
Preparing and mixing your SOMAVERT medicine
The SOMAVERT medicine comes as a dry powder. Before you use SOMAVERT, you must mix the dry powder with the vial of diluent that comes in the SOMAVERT package.
Do not use any other liquid to mix the medicine.
Note: If you need to give 2 injections for your SOMAVERT dose, you need 2 packages of SOMAVERT to prepare 2 separate vials of medicine.
Step 1. Remove 1 package of SOMAVERT from the refrigerator about 10 minutes before you plan to give your SOMAVERT injection. Let the SOMAVERT stand at room temperature to warm up the medicine.
Step 2. Wash your hands with soap and warm water. Dry your hands well.
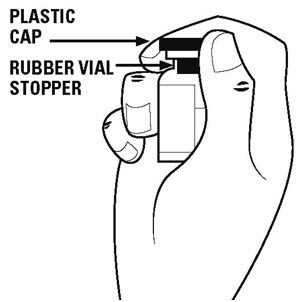
Step 3. Remove the plastic caps from the tops of the powder vial and the diluent vial. See Figure B.
Do not touch the rubber vial stoppers. The stoppers are clean. If the stoppers are touched by anything, you must clean them with an alcohol swab before use.
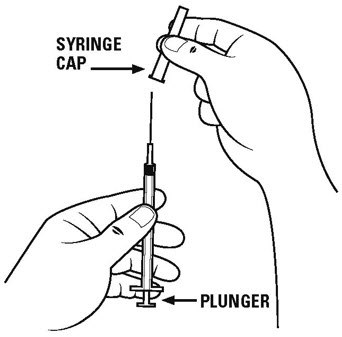
Step 4. Carefully remove the cap from the diluent syringe with the larger needle and set the cap aside on the table. See Figure C.
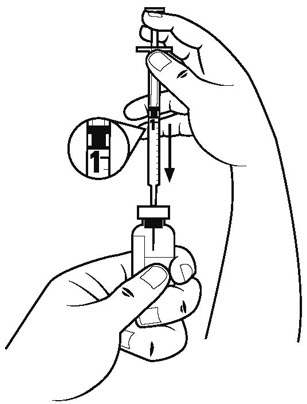
Step 5. Pull the plunger of the diluent syringe out to the 1 mL mark. With 1 hand, firmly hold the vial of diluent. With the other hand, push the needle of the diluent syringe straight through the center of the rubber stopper and deep into the vial. Gently push the plunger in until the air is injected into the vial. See Figure D.
Step 6. Firmly hold the diluent vial and syringe together, with the needle still in the vial. Then turn the diluent vial with the diluent syringe upside down. Hold them at eye level. See Figure E.
Step 7. Slide 1 hand carefully down the diluent vial so you can firmly hold the neck of the vial with your thumb and forefinger. Hold the upper part of the syringe with your other fingers. With the other hand, slowly pull the plunger out to the 1 mL mark on the diluent syringe. See Figure F.
Step 8. Check the diluent syringe for air bubbles. If you see bubbles, tap the diluent syringe barrel until the bubbles rise to the top of the syringe. Carefully push the plunger in to push only the air bubbles back into the vial. See Figure G. If you push too much of the liquid back into the vial, pull the plunger out again to the 1 mL mark.
Step 9. Make sure that 1 mL of diluent remains in the diluent syringe. Pull the needle out of the vial. The vial should still have diluent in it. Do not use the leftover diluent in the vial.
Step 10. Push the needle of the diluent syringe straight through the stopper of the vial of powdered SOMAVERT.
Then, tilt the diluent syringe to the side and gently push the plunger to inject the diluent down the inner side of the SOMAVERT powder vial. Be sure the diluent does not fall directly on the powder, but flows down the inside wall of the vial. See Figure H.
Step 11. When the diluent syringe is empty, pull the needle out of the powder vial.
Throw away the diluent vial with the leftover liquid in it. Throw away the diluent syringe and needle in the sharps container. See "Disposing of used needles and syringes" at the end of these instructions.
Step 12. Hold the medicine vial of SOMAVERT upright between your hands and gently roll it to dissolve the powder into a solution. See Figure I.
- •
- Do not shake the medicine vial. Shaking may destroy the medicine.
- •
- The liquid medicine should be clear after the powder is dissolved. Do not use the vial if:
- o
- the liquid medicine looks cloudy, hazy, or slightly colored or
- o
- you see solid particles in the liquid medicine or
- o
- you see foam in the vial
- Tell your pharmacist and ask for another vial. Do not throw the vial away because the pharmacist may ask that you return it.
- •
- Inject SOMAVERT within 6 hours of mixing it. If you wait more than 6 hours, you must throw away the medicine without injecting it.
- •
- Each mixed medicine vial contains 1 dose of SOMAVERT. Do not split the liquid medicine into multiple doses.
Preparing your SOMAVERT injection syringe:
Step 13. Clean the rubber stopper of the vial of SOMAVERT with an alcohol swab.
- •
- Carefully remove the cap from the insulin syringe and set the cap on the table.
- •
- Pull the insulin syringe plunger out to the 1 mL mark. With 1 hand, firmly hold the vial. With the other hand, push the needle straight through the center of the rubber stopper and deep into the vial. Gently push the plunger in until the air is injected into the medicine vial. See Figure J.
Step 14. Firmly hold the medicine solution vial and insulin syringe together, with the needle still deeply inserted into the vial. Carefully turn the vial and syringe together upside down. Hold them at eye level. See Figure K.
Step 15. Slide 1 hand carefully down the medicine solution vial so you can firmly hold the neck of the vial with your thumb and forefinger. Hold the upper part of the syringe with your other fingers. With the other hand, slowly pull the plunger out to the 1 mL mark on the insulin syringe. See Figure L.
Step 16. Check the insulin syringe for air bubbles. If you see bubbles, tap the insulin syringe barrel until the bubbles rise to the top of the syringe. Carefully push the plunger in to push only the air bubbles back into the vial. See Figure M.
Step 17. Withdraw the entire 1 mL of medicine solution from the vial. If your dose of SOMAVERT is less than 1 mL, your healthcare provider will tell you how much medicine solution to withdraw. Slowly withdraw the needle to keep the tip in the liquid until you get all the medicine solution you need out of the vial. See Figure N.
Note:
- •
- If your dose of SOMAVERT is less than 1 mL, your healthcare provider will tell you how much medicine solution to withdraw.
- •
- If your dose of SOMAVERT is more than 1 mL, your healthcare provider will tell you how much more medicine solution to withdraw from a second vial into another syringe, and where to give your second injection.
Set the syringe and needle on the table without anything touching the needle.
Selecting your SOMAVERT injection site:
Step 18. SOMAVERT is injected under the skin (subcutaneous). Injection sites may include your upper arm, upper thigh, stomach area (abdomen) and buttocks. See Figure O.
- •
- Choose your injection site from 1 of the areas your healthcare provider told you to use. Only a caregiver can inject in your upper arm.
- •
- Choose a different injection site each day so lumps do not develop in your skin. Keep a record of each day's injection site as you inject your daily dose of SOMAVERT.
- •
- Do not use an area of your body that has:
- o
- a rash
- o
- broken skin
- o
- bruising
- o
- lumps in your skin
- •
- If you need to give 2 injections for your dose of SOMAVERT, choose a different site for your second injection.
Giving your SOMAVERT injection:
Step 19. Clean your injection site with an alcohol swab. See Figure P.
Let your skin dry before you inject your medicine.
Step 20. With 1 hand, gently pinch up your skin at your injection site. See Figure Q.
Step 21. Carefully pick up the insulin syringe with your other hand and hold it like a pen. In a single, smooth motion, push the needle straight down and completely into your skin (at a 90-degree angle).
- •
- Keep the needle pushed all the way into your skin while you slowly push the syringe plunger in with the index finger of your other hand. See Figure R.
- •
- Keep the needle all the way into your skin until all of the medicine is injected under your skin and the insulin syringe is empty.
Step 22. Release your pinched skin and pull the needle straight out. See Figure S.
Step 23. Do not rub your injection area. A small amount of bleeding may happen.
If you have a small amount of bleeding, press a small clean, dry cotton pad over the area and press gently for 1 or 2 minutes, or until the bleeding has stopped. See Figure T.
Disposing of used needles and syringes:
- •
- Put your used needles and syringes in a FDA-cleared sharps disposal container right away after use. See Figure U. Do not throw away (dispose of) loose needles and syringes in your household trash.
- •
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- o
- made of a heavy-duty plastic,
- o
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- o
- upright and stable during use,
- o
- leak-resistant, and
- o
- properly labeled to warn of hazardous waste inside the container.
- •
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpdisposal.
- •
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.

U.S. License No. 1216
LAB-1362-3.0
Revised: July 2023
Other

U.S. License No. 1216
This product's labeling may have been updated. For the most recent prescribing information, please visit www.pfizer.com.
LAB-0196-23.0
What is SOMAVERT?
What is SOMAVERT?
Before you use SOMAVERT, tell your healthcare provider about all of your medical conditions, including if you:
- •
- are allergic to pegvisomant or any of the ingredients in SOMAVERT. Do not take SOMAVERT if you are allergic to pegvisomant or any of the ingredients in SOMAVERT. See the end of this Patient Information leaflet for a complete list of ingredients in SOMAVERT.
- •
- have diabetes.
- •
- have or have had liver problems.
- •
- are pregnant or plan to become pregnant. It is not known if SOMAVERT will harm your unborn baby. Tell your healthcare provider if you become pregnant while using SOMAVERT.
- •
- are breastfeeding or plan to breastfeed. It is not known if SOMAVERT passes into your breast milk. You and your healthcare provider should decide how you will feed your baby if you take SOMAVERT.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
SOMAVERT may affect the way other medicines work, and other medicines may affect how SOMAVERT works. Especially tell your healthcare provider if you take:
- •
- insulin or other medicines used to treat diabetes.
- •
- narcotics (opioid medicines). Your healthcare provider may change your dose of SOMAVERT if you take opioids.
If you are not sure, ask your healthcare provider or pharmacist whether you take these medicines.
How should I use SOMAVERT?
How should I use SOMAVERT?
- •
- Read the Instructions for Use at the end of this Patient Information for information about the right way to use SOMAVERT.
- •
- Your healthcare provider should do blood tests to check your liver and insulin-like growth factor-1 (IGF-1) levels before you start and while you use SOMAVERT. Your healthcare provider may need to change your dose of SOMAVERT.
- •
- SOMAVERT is given 1 time each day as an injection under your skin (subcutaneous). Some people may need to give 2 injections for their dose each day. Your healthcare provider will tell you if you need to give 2 injections for your dose.
- •
- Your first injection of SOMAVERT should be given by your healthcare provider.
- •
- Your healthcare provider will teach you or your caregiver how to use SOMAVERT.
- •
- If you use too much SOMAVERT, call your healthcare provider right away.
- •
- If you miss a dose of SOMAVERT, just take the next dose at the regular time. Do not take 2 doses at the same time. If you are not sure about your dosing, ask your healthcare provider.
What are the possible side effects of SOMAVERT?
What are the possible side effects of SOMAVERT?
SOMAVERT may cause serious side effects, including:- •
- changes in your blood sugar level. Your healthcare provider may change your dose of diabetes medicine while you take SOMAVERT.
- •
- liver problems. Stop injecting SOMAVERT right away and call your healthcare provider if you have any of the following symptoms of liver problems:
- o
- yellowing of your eyes (jaundice)
- o
- dark, amber-colored urine
- o
- feeling very tired (fatigue or exhaustion)
- o
- nausea and vomiting
- o
- pain in your stomach (abdomen)
- o
- generalized swelling
- o
- bruising easily
- •
- skin thickening at your injection site that could lead to lumps (lipohypertrophy)
- •
- allergic reactions. Call your healthcare provider right away if you have any of the following symptoms of a serious allergic reaction:
- o
- swelling of your face, tongue, lips, or throat
- o
- wheezing or trouble breathing
- o
- skin rash, redness, or swelling
- o
- severe itching
- o
- dizziness or fainting
The most common side effects of SOMAVERT include:
- •
- pain
- •
- infection
- •
- nausea
- •
- flu syndrome
- •
- injection site reaction
- •
- diarrhea
- •
- abnormal liver tests. If your liver test results are too high, you may have to have more frequent liver tests
These are not all of the possible side effects of SOMAVERT. For more information, ask your healthcare provider or pharmacist.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store SOMAVERT?
How should I store SOMAVERT?
- •
- Before you mix the SOMAVERT powder and the liquid:
- o
- Store SOMAVERT in a refrigerator between 36°F to 46°F (2°C to 8°C).
- o
- Do not freeze SOMAVERT.
- •
- After you mix the SOMAVERT powder and liquid:
- o
- Keep the mixed SOMAVERT at room temperature between 59°F to 77°F (15°C to 25°C).
- o
- Keep SOMAVERT inside the vial or the syringe until you are ready to inject it.
- o
- You must use the mixed SOMAVERT within 6 hours after you mix it.
- o
- If you have not used the mixed SOMAVERT within 6 hours, throw the SOMAVERT away.
Keep SOMAVERT and all medicines out of the reach of children.
General information about the safe and effective use of SOMAVERT.
General information about the safe and effective use of SOMAVERT.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use SOMAVERT for a condition for which it was not prescribed. Do not give SOMAVERT to other people, even if they have the same symptoms that you have. It may harm them.
This Patient Information summarizes the most important information about SOMAVERT. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about SOMAVERT that is written for health professionals.
What are the ingredients in SOMAVERT?
What are the ingredients in SOMAVERT?
Active ingredient: pegvisomant, including polyethylene glycolInactive ingredients: glycine, mannitol, sodium dihydrogen phosphate monohydrate, and sodium phosphate dibasic anhydrous.
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
SOMAVERT (SOM-ah-vert)
(pegvisomant)
for injection, for subcutaneous use
Read this Instructions for Use before you start using SOMAVERT and each time you get a refill. There may be new information. This leaflet does not take the place of talking to your healthcare provider about your medical condition or your treatment. Your healthcare provider should show you or a caregiver how to inject SOMAVERT the right way before you inject it for the first time.
Important Information:
- •
- SOMAVERT is for use under the skin only (subcutaneous).
- •
- Do not share your SOMAVERT syringes or needles with anyone else. You may give an infection to them or get an infection from them.
- •
- Do not use SOMAVERT after the expiration date (EXP) on the label.
- •
- Inject SOMAVERT within 6 hours of mixing it. If you wait more than 6 hours, you must throw away the medicine without injecting it.
Supplies you need to give each injection of SOMAVERT. See Figure A.
- •
- 1 package of SOMAVERT that contains:
- o
- 1 vial of powdered SOMAVERT medicine (powder vial)
- o
- 1 vial of liquid (diluent) labeled "Sterile Water for Injection, USP" to mix the powdered medicine
- The vials in the package of SOMAVERT have stoppers that are not made with natural rubber latex.
- The SOMAVERT package does not come with syringes and needles.
- •
- a 1 mL syringe with a 21 gauge to 27 gauge needle that is at least 1 inch long. This is the syringe and needle needed to mix the medicine (diluent syringe).
- •
- a 1 mL insulin syringe with attached needle. This is the syringe needed for your injection.
- •
- 2 alcohol swabs
- •
- 1 small clean, dry cotton pad
- •
- 1 sharps disposal container for throwing away used needles and syringes. See "Disposing of used needles and syringes" at the end of these instructions.
- •
- a clean, flat surface to work on, like a table
Preparing and mixing your SOMAVERT medicine
The SOMAVERT medicine comes as a dry powder. Before you use SOMAVERT, you must mix the dry powder with the vial of diluent that comes in the SOMAVERT package.
Do not use any other liquid to mix the medicine.
Note: If you need to give 2 injections for your SOMAVERT dose, you need 2 packages of SOMAVERT to prepare 2 separate vials of medicine.
Step 1. Remove 1 package of SOMAVERT from the refrigerator about 10 minutes before you plan to give your SOMAVERT injection. Let the SOMAVERT stand at room temperature to warm up the medicine.
Step 2. Wash your hands with soap and warm water. Dry your hands well.
Step 3. Remove the plastic caps from the tops of the powder vial and the diluent vial. See Figure B.
Do not touch the rubber vial stoppers. The stoppers are clean. If the stoppers are touched by anything, you must clean them with an alcohol swab before use.
Step 4. Carefully remove the cap from the diluent syringe with the larger needle and set the cap aside on the table. See Figure C.
Step 5. Pull the plunger of the diluent syringe out to the 1 mL mark. With 1 hand, firmly hold the vial of diluent. With the other hand, push the needle of the diluent syringe straight through the center of the rubber stopper and deep into the vial. Gently push the plunger in until the air is injected into the vial. See Figure D.
Step 6. Firmly hold the diluent vial and syringe together, with the needle still in the vial. Then turn the diluent vial with the diluent syringe upside down. Hold them at eye level. See Figure E.
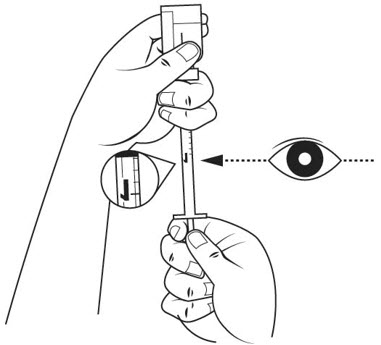
Step 7. Slide 1 hand carefully down the diluent vial so you can firmly hold the neck of the vial with your thumb and forefinger. Hold the upper part of the syringe with your other fingers. With the other hand, slowly pull the plunger out to the 1 mL mark on the diluent syringe. See Figure F.
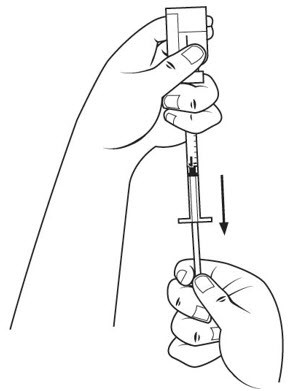
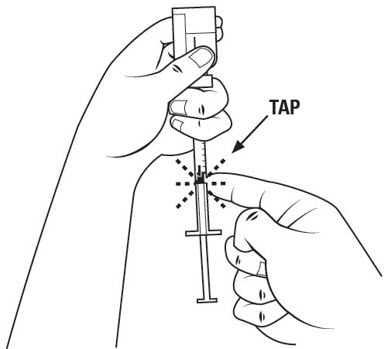
Step 8. Check the diluent syringe for air bubbles. If you see bubbles, tap the diluent syringe barrel until the bubbles rise to the top of the syringe. Carefully push the plunger in to push only the air bubbles back into the vial. See Figure G. If you push too much of the liquid back into the vial, pull the plunger out again to the 1 mL mark.
Step 9. Make sure that 1 mL of diluent remains in the diluent syringe. Pull the needle out of the vial. The vial should still have diluent in it. Do not use the leftover diluent in the vial.
Step 10. Push the needle of the diluent syringe straight through the stopper of the vial of powdered SOMAVERT.
Then, tilt the diluent syringe to the side and gently push the plunger to inject the diluent down the inner side of the SOMAVERT powder vial. Be sure the diluent does not fall directly on the powder, but flows down the inside wall of the vial. See Figure H.
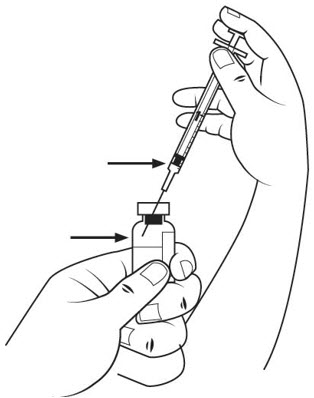
Step 11. When the diluent syringe is empty, pull the needle out of the powder vial.
Throw away the diluent vial with the leftover liquid in it. Throw away the diluent syringe and needle in the sharps container. See "Disposing of used needles and syringes" at the end of these instructions.
Step 12. Hold the medicine vial of SOMAVERT upright between your hands and gently roll it to dissolve the powder into a solution. See Figure I.
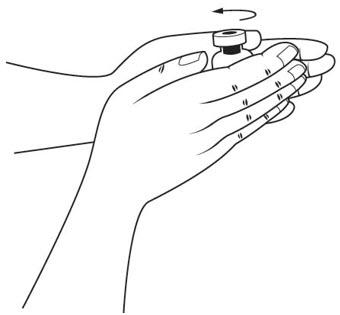
- •
- Do not shake the medicine vial. Shaking may destroy the medicine.
- •
- The liquid medicine should be clear after the powder is dissolved. Do not use the vial if:
- o
- the liquid medicine looks cloudy, hazy, or slightly colored or
- o
- you see solid particles in the liquid medicine or
- o
- you see foam in the vial
- Tell your pharmacist and ask for another vial. Do not throw the vial away because the pharmacist may ask that you return it.
- •
- Inject SOMAVERT within 6 hours of mixing it. If you wait more than 6 hours, you must throw away the medicine without injecting it.
- •
- Each mixed medicine vial contains 1 dose of SOMAVERT. Do not split the liquid medicine into multiple doses.
Preparing your SOMAVERT injection syringe:
Step 13. Clean the rubber stopper of the vial of SOMAVERT with an alcohol swab.
- •
- Carefully remove the cap from the insulin syringe and set the cap on the table.
- •
- Pull the insulin syringe plunger out to the 1 mL mark. With 1 hand, firmly hold the vial. With the other hand, push the needle straight through the center of the rubber stopper and deep into the vial. Gently push the plunger in until the air is injected into the medicine vial. See Figure J.
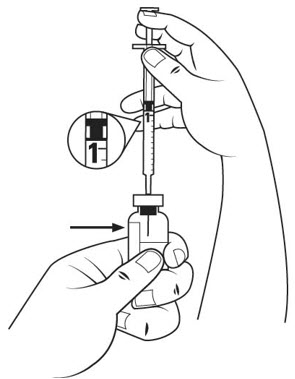
Step 14. Firmly hold the medicine solution vial and insulin syringe together, with the needle still deeply inserted into the vial. Carefully turn the vial and syringe together upside down. Hold them at eye level. See Figure K.
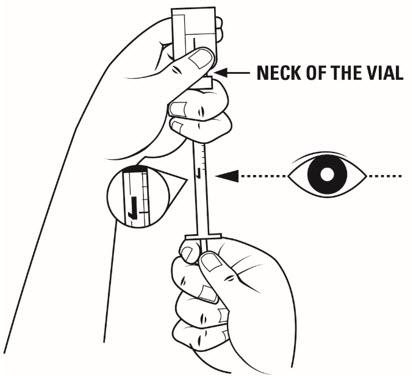
Step 15. Slide 1 hand carefully down the medicine solution vial so you can firmly hold the neck of the vial with your thumb and forefinger. Hold the upper part of the syringe with your other fingers. With the other hand, slowly pull the plunger out to the 1 mL mark on the insulin syringe. See Figure L.
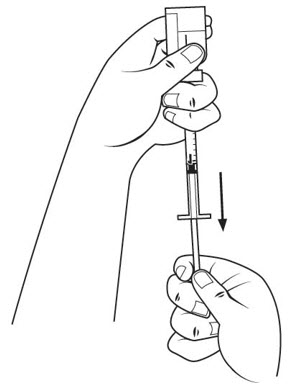
Step 16. Check the insulin syringe for air bubbles. If you see bubbles, tap the insulin syringe barrel until the bubbles rise to the top of the syringe. Carefully push the plunger in to push only the air bubbles back into the vial. See Figure M.
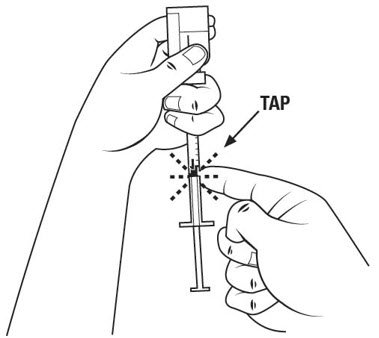
Step 17. Withdraw the entire 1 mL of medicine solution from the vial. If your dose of SOMAVERT is less than 1 mL, your healthcare provider will tell you how much medicine solution to withdraw. Slowly withdraw the needle to keep the tip in the liquid until you get all the medicine solution you need out of the vial. See Figure N.
- •
- If your dose of SOMAVERT is less than 1 mL, your healthcare provider will tell you how much medicine solution to withdraw.
- •
- If your dose of SOMAVERT is more than 1 mL, your healthcare provider will tell you how much more medicine solution to withdraw from a second vial into another syringe, and where to give your second injection.
Set the syringe and needle on the table without anything touching the needle.
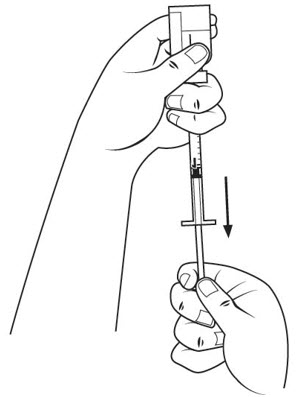
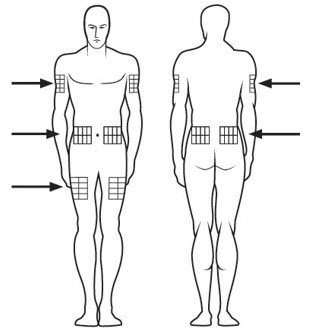
Selecting your SOMAVERT injection site:
Step 18. SOMAVERT is injected under the skin (subcutaneous). Injection sites may include your upper arm, upper thigh, stomach area (abdomen) and buttocks. See Figure O.
- •
- Choose your injection site from 1 of the areas your healthcare provider told you to use. Only a caregiver can inject in your upper arm.
- •
- Choose a different injection site each day so lumps do not develop in your skin. Keep a record of each day's injection site as you inject your daily dose of SOMAVERT.
- •
- Do not use an area of your body that has:
- o
- a rash
- o
- broken skin
- o
- bruising
- o
- lumps in your skin
- •
- If you need to give 2 injections for your dose of SOMAVERT, choose a different site for your second injection.
Giving your SOMAVERT injection:
Step 19. Clean your injection site with an alcohol swab. See Figure P.
Let your skin dry before you inject your medicine.
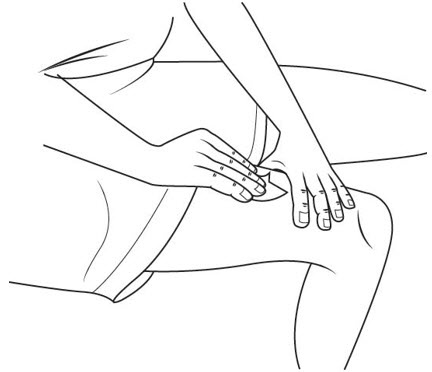
Step 20. With 1 hand, gently pinch up your skin at your injection site. See Figure Q.
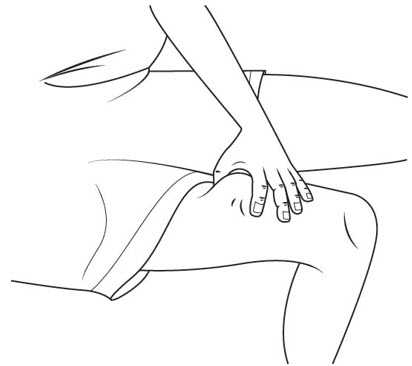
Step 21. Carefully pick up the insulin syringe with your other hand and hold it like a pen. In a single, smooth motion, push the needle straight down and completely into your skin (at a 90-degree angle).
- •
- Keep the needle pushed all the way into your skin while you slowly push the syringe plunger in with the index finger of your other hand. See Figure R.
- •
- Keep the needle all the way into your skin until all of the medicine is injected under your skin and the insulin syringe is empty.
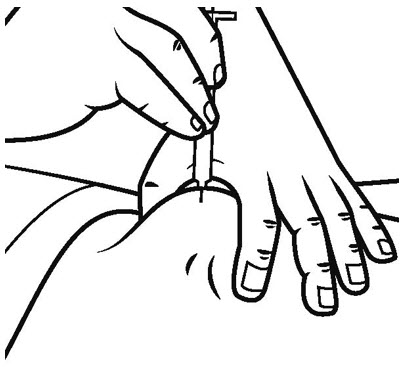
Step 22. Release your pinched skin and pull the needle straight out. See Figure S.
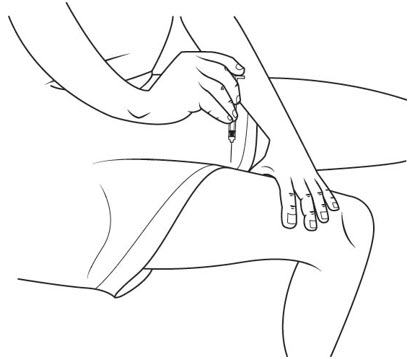
Step 23. Do not rub your injection area. A small amount of bleeding may happen.
If you have a small amount of bleeding, press a small clean, dry cotton pad over the area and press gently for 1 or 2 minutes, or until the bleeding has stopped. See Figure T.
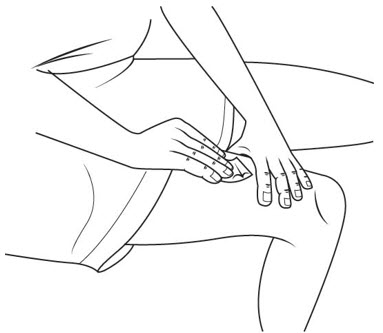
Disposing of used needles and syringes:
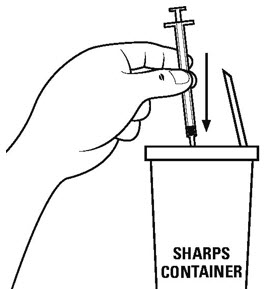
- •
- Put your used needles and syringes in a FDA-cleared sharps disposal container right away after use. See Figure U. Do not throw away (dispose of) loose needles and syringes in your household trash.
- •
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- o
- made of a heavy-duty plastic,
- o
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- o
- upright and stable during use,
- o
- leak-resistant, and
- o
- properly labeled to warn of hazardous waste inside the container.
- •
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpdisposal.
- •
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
U.S. License No. 1216
LAB-1362-3.0
Revised: July 2023
Full Patient Information
Full Patient Information
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Inform patients (and/or their caregivers) of the following information to aid in the safe and effective use of SOMAVERT:
- •
- Not to use SOMAVERT if they are allergic to SOMAVERT or anything in it.
- •
- They will need blood testing to check IGF-1 levels and liver tests before and during treatment with SOMAVERT and that the dose of SOMAVERT may be changed based on the results of these tests.
- •
- SOMAVERT has not been studied in pregnant women and instruct them to notify their healthcare provider as soon as they are aware that they are pregnant.
- •
- It is not known whether SOMAVERT is excreted in human milk and instruct them to notify their healthcare provider if they plan to do so.
- •
- Pregnancy: Inform female patients that treatment with SOMAVERT may result in unintended pregnancy [see Females and Males of Reproductive Potential (8.3)].
Advise patients (and/or their caregivers) of the following adverse reactions:
- •
- The most common reported adverse reactions are injection site reaction, elevations of liver tests, pain, nausea, and diarrhea.
- •
- If they have liver test elevations they may need to have more frequent liver tests and/or discontinue SOMAVERT. Instruct patients to immediately discontinue therapy and contact their physician if they become jaundiced.
- •
- GH-secreting tumors may enlarge in people with acromegaly and that these tumors need to be watched carefully and monitored by MRI imaging.
- •
- Thickening under the skin may occur at the injection site that could lead to lumps and that switching sites may prevent or lessen this.
- •
- If they have diabetes mellitus, they may require careful monitoring and dose reductions of insulin and/or oral hypoglycemic agents while on SOMAVERT.
- •
- If they take opioids, they may need higher SOMAVERT doses to achieve appropriate IGF-1 suppression.
Advise patients that SOMAVERT is supplied as lyophilized powder in different strengths of 10 mg, 15 mg, 20 mg, 25 mg, and 30 mg in a sterile glass vial within a package also containing a single-dose flip top vial of sterile water (diluent) for injection. Advise patients that the stoppers on both vials are not made with natural rubber latex. Advise patients to follow the directions for reconstitution provided with each package including shaking may cause denaturation (destruction) of the active ingredient (therefore do not shake).
Advise patients that the package of SOMAVERT should be stored in a refrigerator 2°C to 8°C (36°F to 46°F) prior to use. It should NOT BE FROZEN.
INSTRUCTIONS FOR USE
SOMAVERT (SOM-ah-vert)
(pegvisomant)
for injection, for subcutaneous use
Read this Instructions for Use before you start using SOMAVERT and each time you get a refill. There may be new information. This leaflet does not take the place of talking to your healthcare provider about your medical condition or your treatment. Your healthcare provider should show you or a caregiver how to inject SOMAVERT the right way before you inject it for the first time.
Important Information:
- •
- SOMAVERT is for use under the skin only (subcutaneous).
- •
- Do not share your SOMAVERT syringes or needles with anyone else. You may give an infection to them or get an infection from them.
- •
- Do not use SOMAVERT after the expiration date (EXP) on the label.
- •
- Inject SOMAVERT within 6 hours of mixing it. If you wait more than 6 hours, you must throw away the medicine without injecting it.
Supplies you need to give each injection of SOMAVERT. See Figure A.
- •
- 1 package of SOMAVERT that contains:
- o
- 1 vial of powdered SOMAVERT medicine (powder vial)
- o
- 1 vial of liquid (diluent) labeled "Sterile Water for Injection, USP" to mix the powdered medicine
- The vials in the package of SOMAVERT have stoppers that are not made with natural rubber latex.
- The SOMAVERT package does not come with syringes and needles.
- •
- a 1 mL syringe with a 21 gauge to 27 gauge needle that is at least 1 inch long. This is the syringe and needle needed to mix the medicine (diluent syringe).
- •
- a 1 mL insulin syringe with attached needle. This is the syringe needed for your injection.
- •
- 2 alcohol swabs
- •
- 1 small clean, dry cotton pad
- •
- 1 sharps disposal container for throwing away used needles and syringes. See "Disposing of used needles and syringes" at the end of these instructions.
- •
- a clean, flat surface to work on, like a table
Preparing and mixing your SOMAVERT medicine
The SOMAVERT medicine comes as a dry powder. Before you use SOMAVERT, you must mix the dry powder with the vial of diluent that comes in the SOMAVERT package.
Do not use any other liquid to mix the medicine.
Note: If you need to give 2 injections for your SOMAVERT dose, you need 2 packages of SOMAVERT to prepare 2 separate vials of medicine.
Step 1. Remove 1 package of SOMAVERT from the refrigerator about 10 minutes before you plan to give your SOMAVERT injection. Let the SOMAVERT stand at room temperature to warm up the medicine.
Step 2. Wash your hands with soap and warm water. Dry your hands well.
Step 3. Remove the plastic caps from the tops of the powder vial and the diluent vial. See Figure B.
Do not touch the rubber vial stoppers. The stoppers are clean. If the stoppers are touched by anything, you must clean them with an alcohol swab before use.
Step 4. Carefully remove the cap from the diluent syringe with the larger needle and set the cap aside on the table. See Figure C.
Step 5. Pull the plunger of the diluent syringe out to the 1 mL mark. With 1 hand, firmly hold the vial of diluent. With the other hand, push the needle of the diluent syringe straight through the center of the rubber stopper and deep into the vial. Gently push the plunger in until the air is injected into the vial. See Figure D.
Step 6. Firmly hold the diluent vial and syringe together, with the needle still in the vial. Then turn the diluent vial with the diluent syringe upside down. Hold them at eye level. See Figure E.
Step 7. Slide 1 hand carefully down the diluent vial so you can firmly hold the neck of the vial with your thumb and forefinger. Hold the upper part of the syringe with your other fingers. With the other hand, slowly pull the plunger out to the 1 mL mark on the diluent syringe. See Figure F.
Step 8. Check the diluent syringe for air bubbles. If you see bubbles, tap the diluent syringe barrel until the bubbles rise to the top of the syringe. Carefully push the plunger in to push only the air bubbles back into the vial. See Figure G. If you push too much of the liquid back into the vial, pull the plunger out again to the 1 mL mark.
Step 9. Make sure that 1 mL of diluent remains in the diluent syringe. Pull the needle out of the vial. The vial should still have diluent in it. Do not use the leftover diluent in the vial.
Step 10. Push the needle of the diluent syringe straight through the stopper of the vial of powdered SOMAVERT.
Then, tilt the diluent syringe to the side and gently push the plunger to inject the diluent down the inner side of the SOMAVERT powder vial. Be sure the diluent does not fall directly on the powder, but flows down the inside wall of the vial. See Figure H.
Step 11. When the diluent syringe is empty, pull the needle out of the powder vial.
Throw away the diluent vial with the leftover liquid in it. Throw away the diluent syringe and needle in the sharps container. See "Disposing of used needles and syringes" at the end of these instructions.
Step 12. Hold the medicine vial of SOMAVERT upright between your hands and gently roll it to dissolve the powder into a solution. See Figure I.
- •
- Do not shake the medicine vial. Shaking may destroy the medicine.
- •
- The liquid medicine should be clear after the powder is dissolved. Do not use the vial if:
- o
- the liquid medicine looks cloudy, hazy, or slightly colored or
- o
- you see solid particles in the liquid medicine or
- o
- you see foam in the vial
- Tell your pharmacist and ask for another vial. Do not throw the vial away because the pharmacist may ask that you return it.
- •
- Inject SOMAVERT within 6 hours of mixing it. If you wait more than 6 hours, you must throw away the medicine without injecting it.
- •
- Each mixed medicine vial contains 1 dose of SOMAVERT. Do not split the liquid medicine into multiple doses.
Preparing your SOMAVERT injection syringe:
Step 13. Clean the rubber stopper of the vial of SOMAVERT with an alcohol swab.
- •
- Carefully remove the cap from the insulin syringe and set the cap on the table.
- •
- Pull the insulin syringe plunger out to the 1 mL mark. With 1 hand, firmly hold the vial. With the other hand, push the needle straight through the center of the rubber stopper and deep into the vial. Gently push the plunger in until the air is injected into the medicine vial. See Figure J.
Step 14. Firmly hold the medicine solution vial and insulin syringe together, with the needle still deeply inserted into the vial. Carefully turn the vial and syringe together upside down. Hold them at eye level. See Figure K.
Step 15. Slide 1 hand carefully down the medicine solution vial so you can firmly hold the neck of the vial with your thumb and forefinger. Hold the upper part of the syringe with your other fingers. With the other hand, slowly pull the plunger out to the 1 mL mark on the insulin syringe. See Figure L.
Step 16. Check the insulin syringe for air bubbles. If you see bubbles, tap the insulin syringe barrel until the bubbles rise to the top of the syringe. Carefully push the plunger in to push only the air bubbles back into the vial. See Figure M.
Step 17. Withdraw the entire 1 mL of medicine solution from the vial. If your dose of SOMAVERT is less than 1 mL, your healthcare provider will tell you how much medicine solution to withdraw. Slowly withdraw the needle to keep the tip in the liquid until you get all the medicine solution you need out of the vial. See Figure N.
Note:
- •
- If your dose of SOMAVERT is less than 1 mL, your healthcare provider will tell you how much medicine solution to withdraw.
- •
- If your dose of SOMAVERT is more than 1 mL, your healthcare provider will tell you how much more medicine solution to withdraw from a second vial into another syringe, and where to give your second injection.
Set the syringe and needle on the table without anything touching the needle.
Selecting your SOMAVERT injection site:
Step 18. SOMAVERT is injected under the skin (subcutaneous). Injection sites may include your upper arm, upper thigh, stomach area (abdomen) and buttocks. See Figure O.
- •
- Choose your injection site from 1 of the areas your healthcare provider told you to use. Only a caregiver can inject in your upper arm.
- •
- Choose a different injection site each day so lumps do not develop in your skin. Keep a record of each day's injection site as you inject your daily dose of SOMAVERT.
- •
- Do not use an area of your body that has:
- o
- a rash
- o
- broken skin
- o
- bruising
- o
- lumps in your skin
- •
- If you need to give 2 injections for your dose of SOMAVERT, choose a different site for your second injection.
Giving your SOMAVERT injection:
Step 19. Clean your injection site with an alcohol swab. See Figure P.
Let your skin dry before you inject your medicine.
Step 20. With 1 hand, gently pinch up your skin at your injection site. See Figure Q.
Step 21. Carefully pick up the insulin syringe with your other hand and hold it like a pen. In a single, smooth motion, push the needle straight down and completely into your skin (at a 90-degree angle).
- •
- Keep the needle pushed all the way into your skin while you slowly push the syringe plunger in with the index finger of your other hand. See Figure R.
- •
- Keep the needle all the way into your skin until all of the medicine is injected under your skin and the insulin syringe is empty.
Step 22. Release your pinched skin and pull the needle straight out. See Figure S.
Step 23. Do not rub your injection area. A small amount of bleeding may happen.
If you have a small amount of bleeding, press a small clean, dry cotton pad over the area and press gently for 1 or 2 minutes, or until the bleeding has stopped. See Figure T.
Disposing of used needles and syringes:
- •
- Put your used needles and syringes in a FDA-cleared sharps disposal container right away after use. See Figure U. Do not throw away (dispose of) loose needles and syringes in your household trash.
- •
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- o
- made of a heavy-duty plastic,
- o
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- o
- upright and stable during use,
- o
- leak-resistant, and
- o
- properly labeled to warn of hazardous waste inside the container.
- •
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpdisposal.
- •
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
U.S. License No. 1216
LAB-1362-3.0
Revised: July 2023
Resources
Didn’t find what you were looking for?
Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
Pfizer Safety
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this product, click the link below to submit your information: Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer product, please call Pfizer Medical Information at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800)-332-1088.