ZYVOX®
(linezolid)
Find ZYVOX® medical information:
Find ZYVOX® medical information:
ZYVOX® Quick Finder
17 PATIENT COUNSELING INFORMATION
17 PATIENT COUNSELING INFORMATION
Important Administration Instructions
Advise patients that ZYVOX may be taken with or without food.
Peripheral and Optic Neuropathy
Advise patients to inform their physician if they experience changes in vision while taking ZYVOX [see Warnings and Precautions (5.2)].
Serotonin Syndrome
Advise patients to inform their physician if taking serotonergic agents, including serotonin re-uptake inhibitors or other antidepressants and opioids [see Warnings and Precautions (5.3)].
Potential Interactions Producing Elevation of Blood Pressure
- •
- Advise patients to inform their physician if they have a history of hypertension.
- •
- Advise patients to avoid large quantities of foods or beverages with high tyramine content while taking ZYVOX. Foods high in tyramine content include those that may have undergone protein changes by aging, fermentation, pickling, or smoking to improve flavor, such as aged cheeses, fermented or air-dried meats, sauerkraut, soy sauce, tap beers, and red wines. The tyramine content of any protein-rich food may be increased if stored for long periods or improperly refrigerated.
- •
- Advise patients to inform their physician if taking medications containing pseudoephedrine HCl or phenylpropanolamine HCl, such as cold remedies and decongestants [see Warnings and Precautions (5.6)].
Lactic Acidosis
Advise patients to inform their physician if they experience repeated episodes of nausea or vomiting while receiving ZYVOX [see Warnings and Precautions (5.7)].
Convulsions
Advise patients to inform their physician if they have a history of seizures or convulsions [see Warnings and Precautions (5.8)].
Rhabdomyolysis
Advise patients to inform their physician if they experience signs and symptoms of rhabdomyolysis including muscle pain, tenderness or weakness and dark urine [see Warnings and Precautions (5.9)].
Hypoglycemia
Advise patients to inform their physician if they have diabetes mellitus. Hypoglycemic reactions, such as diaphoresis and tremulousness, along with low blood glucose measurements may occur when treated with linezolid. If such reactions occur, patients should contact a physician or other health professional for proper treatment [see Warnings and Precautions (5.10)].
Hyponatremia and/or SIADH
Advise patients at risk for hyponatremia to inform their physician if they experience signs and symptoms of hyponatremia and/or SIADH, including confusion, somnolence, generalized weakness, and respiratory distress [see Warnings and Precautions (5.11)].
Phenylketonuria
Advise patients with phenylketonuria (PKU) that each 5 mL of the 100 mg/5 mL ZYVOX for Oral Suspension contains 20 mg phenylalanine. The other ZYVOX formulations do not contain phenylalanine. Phenylalanine can be harmful to patients with phenylketonuria. Contact your physician or pharmacist when prescribed with ZYVOX for Oral Suspension [see Warnings and Precautions (5.12)].
Antibacterial Resistance
Patients should be counseled that antibacterial drugs including ZYVOX should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When ZYVOX is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by ZYVOX or other antibacterial drugs in the future.
Diarrhea
Diarrhea is a common problem caused by antibacterial drugs, which usually ends when the antibacterial drug is discontinued. Sometimes after starting treatment with antibacterial drugs, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibacterial drug. If this occurs, patients should contact their physician as soon as possible [see Warnings and Precautions (5.5)].
Infertility
Advise male patients that ZYVOX may reversibly impair fertility [see Use in Specific Populations (8.3)].
Highlights
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ZYVOX safely and effectively. See full prescribing information for ZYVOX. ZYVOX (linezolid) injection, for intravenous use ZYVOX (linezolid) tablets, for oral use ZYVOX (linezolid) for oral suspension Initial U.S. Approval: 2000 RECENT MAJOR CHANGESINDICATIONS AND USAGEZYVOX is an oxazolidinone-class antibacterial indicated in adults and children for the treatment of the following infections caused by susceptible Gram-positive bacteria: Nosocomial pneumonia (1.1); Community-acquired pneumonia (1.2); Complicated skin and skin structure infections, including diabetic foot infections, without concomitant osteomyelitis (1.3); Uncomplicated skin and skin structure infections (1.4); Vancomycin-resistant Enterococcus faecium infections. (1.5)
To reduce the development of drug-resistant bacteria and maintain the effectiveness of ZYVOX formulations and other antibacterial drugs, ZYVOX should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria. (1.7) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSCONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (>5% of adult and/or pediatric patients treated with ZYVOX) include: diarrhea, vomiting, headache, nausea, and anemia. (6) DRUG INTERACTIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 6/2024 | ||||||||||||||||||||||
Indications and Usage
1 INDICATIONS AND USAGE
1.1 Nosocomial Pneumonia
ZYVOX is indicated for the treatment of nosocomial pneumonia caused by Staphylococcus aureus (methicillin-susceptible and -resistant isolates) or Streptococcus pneumoniae [see Clinical Studies (14)].
1.2 Community-acquired Pneumonia
ZYVOX is indicated for the treatment of community-acquired pneumonia caused by Streptococcus pneumoniae, including cases with concurrent bacteremia, or Staphylococcus aureus (methicillin-susceptible isolates only) [see Clinical Studies (14)].
1.3 Complicated Skin and Skin Structure Infections
ZYVOX is indicated for the treatment of complicated skin and skin structure infections, including diabetic foot infections, without concomitant osteomyelitis, caused by Staphylococcus aureus (methicillin-susceptible and -resistant isolates), Streptococcus pyogenes, or Streptococcus agalactiae. ZYVOX has not been studied in the treatment of decubitus ulcers [see Clinical Studies (14)].
1.4 Uncomplicated Skin and Skin Structure Infections
ZYVOX is indicated for the treatment of uncomplicated skin and skin structure infections caused by Staphylococcus aureus (methicillin-susceptible isolates only) or Streptococcus pyogenes [see Clinical Studies (14)].
1.5 Vancomycin-resistant Enterococcus faecium Infections
ZYVOX is indicated for the treatment of vancomycin-resistant Enterococcus faecium infections, including cases with concurrent bacteremia [see Clinical Studies (14)].
1.6 Limitations of Use
- •
- ZYVOX is not indicated for the treatment of Gram-negative infections. It is critical that specific Gram-negative therapy be initiated immediately if a concomitant Gram-negative pathogen is documented or suspected [see Warnings and Precautions (5.4)].
- •
- The safety and efficacy of ZYVOX formulations given for longer than 28 days have not been evaluated in controlled clinical trials [see Clinical Studies (14)].
1.7 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of ZYVOX and other antibacterial drugs, ZYVOX should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
Dosage and Administration
2 DOSAGE AND ADMINISTRATION
2.1 General Dosage and Administration
The recommended dosage for ZYVOX formulations for the treatment of infections is described in Table 1.
| Dosage, Route and Frequency of Administration | Recommended Duration of Treatment (consecutive days) | ||
|---|---|---|---|
| Infection* | Pediatric Patients† (Birth through 11 Years of Age) | Adults and Adolescents (12 Years and Older) | |
| |||
Nosocomial pneumonia | 10 mg/kg intravenously or oral‡ every 8 hours | 600 mg intravenously or oral‡ every 12 hours | 10 to 14 |
Community-acquired pneumonia, including concurrent bacteremia | |||
Complicated skin and skin structure infections | |||
Vancomycin-resistant Enterococcus faecium infections, including concurrent bacteremia | 10 mg/kg intravenously or oral‡ every 8 hours | 600 mg intravenously or oral‡ every 12 hours | 14 to 28 |
Uncomplicated skin and skin structure infections | less than 5 yrs: 10 mg/kg oral‡ every 8 hours | Adults: 400 mg oral‡ every 12 hours | 10 to 14 |
No dose adjustment is necessary when switching from intravenous to oral administration.
2.2 Intravenous Administration
ZYVOX I.V. Injection is supplied in single-dose, ready-to-use infusion bags. Parenteral drug products should be inspected visually for particulate matter prior to administration. Check for minute leaks by firmly squeezing the bag. If leaks are detected, discard the solution, as sterility may be impaired. Keep the infusion bags in the overwrap until ready to use. Store at room temperature. Protect from freezing. ZYVOX I.V. Injection may exhibit a yellow color that can intensify over time without adversely affecting potency.
ZYVOX I.V. Injection should be administered by intravenous infusion over a period of 30 to 120 minutes. Do not use this intravenous infusion bag in series connections. Additives should not be introduced into this solution. If ZYVOX I.V. Injection is to be given concomitantly with another drug, each drug should be given separately in accordance with the recommended dosage and route of administration for each product. Discard unused portion.
If the same intravenous line is used for sequential infusion of several drugs, the line should be flushed before and after infusion of ZYVOX I.V. Injection with an infusion solution compatible with ZYVOX I.V. Injection and with any other drug(s) administered via this common line.
2.3 Compatibilities
Compatible intravenous solutions include 0.9% Sodium Chloride Injection, USP, 5% Dextrose Injection, USP, and Lactated Ringer's Injection, USP.
2.4 Incompatibilities
Physical incompatibilities resulted when ZYVOX I.V. Injection was combined with the following drugs during simulated Y-site administration: amphotericin B, chlorpromazine HCl, diazepam, pentamidine isothionate, erythromycin lactobionate, phenytoin sodium, and trimethoprim-sulfamethoxazole. Additionally, chemical incompatibility resulted when ZYVOX I.V. Injection was combined with ceftriaxone sodium.
2.5 Constitution of Oral Suspension
ZYVOX for Oral Suspension is supplied as a powder/granule for constitution. Gently tap bottle to loosen powder. Add a total of 123 mL distilled water in two portions. After adding the first half, shake vigorously to wet all of the powder. Then add the second half of the water and shake vigorously to obtain a uniform suspension. After constitution, each 5 mL of the suspension contains 100 mg of linezolid. Before using, gently mix by inverting the bottle 3 to 5 times. Do not shake. Store constituted suspension at room temperature. Use within 21 days after constitution.
Dosage Forms and Strengths
3 DOSAGE FORMS AND STRENGTHS
ZYVOX I.V. Injection: 200 mg/100 mL (2 mg/mL) and 600 mg/300 mL (2 mg/mL) linezolid single-dose, ready-to-use flexible plastic infusion bags in a foil laminate overwrap.
ZYVOX 600 mg Tablet:
white, capsule-shaped, film-coated tablet debossed with "ZYV" on one side and "600" on the other
ZYVOX for Oral Suspension: dry, white to off-white, orange-flavored granule/powder. When constituted as directed, each bottle will contain 150 mL of a suspension providing the equivalent of 100 mg of linezolid per each 5 mL.
Contraindications
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
Myelosuppression (including anemia, leukopenia, pancytopenia, and thrombocytopenia) has been reported in patients receiving linezolid. In cases where the outcome is known, when linezolid was discontinued, the affected hematologic parameters have risen toward pretreatment levels. Thrombocytopenia has been reported more often in patients with severe renal impairment, whether or not on dialysis, and in patients with moderate to severe hepatic impairment. Complete blood counts should be monitored weekly in patients who receive linezolid, particularly in those who receive linezolid for longer than two weeks, those with pre-existing myelosuppression, those with severe renal impairment or moderate to severe hepatic impairment, those receiving concomitant drugs that produce bone marrow suppression, or those with a chronic infection who have received previous or concomitant antibacterial drug therapy. Discontinuation of therapy with linezolid should be considered in patients who develop or have worsening myelosuppression [see Adverse Reactions (6.2)].
5.2 Peripheral and Optic Neuropathy
Peripheral and optic neuropathies have been reported in patients treated with ZYVOX, primarily in those patients treated for longer than the maximum recommended duration of 28 days. In cases of optic neuropathy that progressed to loss of vision, patients were treated for extended periods beyond the maximum recommended duration. Visual blurring has been reported in some patients treated with ZYVOX for less than 28 days. Peripheral and optic neuropathy has also been reported in children.
If patients experience symptoms of visual impairment, such as changes in visual acuity, changes in color vision, blurred vision, or visual field defect, prompt ophthalmic evaluation is recommended. Visual function should be monitored in all patients taking ZYVOX for extended periods (≥ 3 months) and in all patients reporting new visual symptoms regardless of length of therapy with ZYVOX. If peripheral or optic neuropathy occurs, the continued use of ZYVOX in these patients should be weighed against the potential risks.
5.3 Serotonin Syndrome
Spontaneous reports of serotonin syndrome including fatal cases associated with the co-administration of ZYVOX and serotonergic agents, including antidepressants such as selective serotonin reuptake inhibitors (SSRIs), have been reported.
Unless clinically appropriate and patients are carefully observed for signs and/or symptoms of serotonin syndrome or neuroleptic malignant syndrome-like (NMS-like) reactions, linezolid should not be administered to patients with carcinoid syndrome and/or patients taking any of the following medications: serotonin re-uptake inhibitors, tricyclic antidepressants, bupropion, buspirone, serotonin 5-HT1 receptor agonists (triptans), and opioids, including meperidine [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
In some cases, a patient already receiving a serotonergic antidepressant or buspirone may require urgent treatment with linezolid. If alternatives to linezolid are not available and the potential benefits of linezolid outweigh the risks of serotonin syndrome or NMS-like reactions, the serotonergic antidepressant should be stopped promptly and linezolid administered. The patient should be monitored for two weeks (five weeks if fluoxetine was taken) or until 24 hours after the last dose of linezolid, whichever comes first. Symptoms of serotonin syndrome or NMS-like reactions include hyperthermia, rigidity, myoclonus, autonomic instability, and mental status changes that include extreme agitation progressing to delirium and coma. The patient should also be monitored for discontinuation symptoms of the antidepressant (see package insert of the specified agent(s) for a description of the associated discontinuation symptoms).
5.4 Mortality Imbalance in an Investigational Study in Patients with Catheter-Related Bloodstream Infections, Including Those with Catheter-site Infections
An imbalance in mortality was seen in patients treated with linezolid relative to vancomycin/dicloxacillin/oxacillin in an open-label study in seriously ill patients with intravascular catheter-related infections [78/363 (21.5%) vs. 58/363 (16.0%); odds ratio 1.426, 95% CI 0.970, 2.098]. While causality has not been established, this observed imbalance occurred primarily in linezolid-treated patients in whom either Gram-negative pathogens, mixed Gram-negative and Gram-positive pathogens, or no pathogen were identified at baseline, but was not seen in patients with Gram-positive infections only.
Linezolid is not approved and should not be used for the treatment of patients with catheter-related bloodstream infections or catheter-site infections.
Linezolid has no clinical activity against Gram-negative pathogens and is not indicated for the treatment of Gram-negative infections. It is critical that specific Gram-negative therapy be initiated immediately if a concomitant Gram-negative pathogen is documented or suspected [see Indications and Usage (1)].
5.5 Clostridioides difficile-Associated Diarrhea
Clostridioides difficile-Associated Diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including ZYVOX, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial drug use.
Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibacterial drug use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibacterial drug treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
5.6 Potential Interactions Producing Elevation of Blood Pressure
Unless patients are monitored for potential increases in blood pressure, linezolid should not be administered to patients with uncontrolled hypertension, pheochromocytoma, thyrotoxicosis and/or patients taking any of the following types of medications: directly and indirectly acting sympathomimetic agents (e.g., pseudoephedrine), vasopressive agents (e.g., epinephrine, norepinephrine), dopaminergic agents (e.g., dopamine, dobutamine) [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
5.7 Lactic Acidosis
Lactic acidosis has been reported with the use of ZYVOX. In reported cases, patients experienced repeated episodes of nausea and vomiting. Patients who develop recurrent nausea or vomiting, unexplained acidosis, or a low bicarbonate level while receiving ZYVOX should receive immediate medical evaluation.
5.8 Convulsions
Convulsions have been reported in patients when treated with linezolid. In some of these cases, a history of seizures or risk factors for seizures was reported.
5.9 Rhabdomyolysis
Rhabdomyolysis has been reported with the use of linezolid, including ZYVOX [see Adverse Reactions (6.2)]. If signs or symptoms of rhabdomyolysis such as muscle pain, tenderness or weakness, dark urine or elevated creatine phosphokinase are observed, discontinue ZYVOX and initiate appropriate therapy.
5.10 Hypoglycemia
Postmarketing cases of symptomatic hypoglycemia have been reported in patients with diabetes mellitus receiving insulin or oral hypoglycemic agents when treated with linezolid, a reversible, nonselective MAO inhibitor. Some MAO inhibitors have been associated with hypoglycemic episodes in diabetic patients receiving insulin or hypoglycemic agents. While a causal relationship between linezolid and hypoglycemia has not been established, diabetic patients should be cautioned of potential hypoglycemic reactions when treated with linezolid.
If hypoglycemia occurs, a decrease in the dose of insulin or oral hypoglycemic agent, or discontinuation of oral hypoglycemic agent, insulin, or linezolid may be required.
5.11 Hyponatremia and/or Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH)
Postmarketing cases of hyponatremia and/or Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH) have been observed in patients treated with linezolid. In reported cases, the signs and symptoms included confusion, somnolence, generalized weakness, and in severe cases led to respiratory failure and even death. Monitor serum sodium levels regularly in the elderly, in patients taking diuretics, and in other patients at risk of hyponatremia and/or SIADH while taking ZYVOX. If signs and symptoms of hyponatremia and/or SIADH occur, discontinue ZYVOX, and institute appropriate supportive measures.
5.12 Risks in Patients with Phenylketonuria
Phenylalanine can be harmful to patients with phenylketonuria (PKU). ZYVOX for Oral Suspension contains phenylalanine, a component of aspartame. Each 5 mL of the 100 mg/5 mL oral suspension contains 20 mg of phenylalanine. Before prescribing ZYVOX for Oral Suspension to a patient with PKU, consider the combined daily amount of phenylalanine from all sources, including ZYVOX for Oral Suspension.
The other ZYVOX formulations do not contain phenylalanine.
Adverse Reactions
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- •
- Myelosuppression [see Warnings and Precautions (5.1)]
- •
- Peripheral and Optic Neuropathy [see Warnings and Precautions (5.2)]
- •
- Serotonin Syndrome [see Warnings and Precautions (5.3)]
- •
- Clostridioides difficile-Associated Diarrhea [see Warnings and Precautions (5.5)]
- •
- Lactic Acidosis [see Warnings and Precautions (5.7)]
- •
- Convulsions [see Warnings and Precautions (5.8)]
- •
- Rhabdomyolysis [see Warnings and Precautions (5.9)]
- •
- Hypoglycemia [see Warnings and Precautions (5.10)]
- •
- Hyponatremia and/or Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH) [see Warnings and Precautions (5.11)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adults
The safety of ZYVOX formulations was evaluated in 2,046 adult patients enrolled in seven Phase 3 comparator-controlled clinical trials, who were treated for up to 28 days.
Of the patients treated for uncomplicated skin and skin structure infections (uSSSIs), 25.4% of ZYVOX-treated and 19.6% of comparator-treated patients experienced at least one drug-related adverse event. For all other indications, 20.4% of ZYVOX -treated and 14.3% of comparator-treated patients experienced at least one drug-related adverse event.
Table 2 shows the incidence of all-causality, treatment-emergent adverse reactions reported in at least 1% of adult patients in these trials by dose of ZYVOX.
| ADVERSE REACTIONS | Uncomplicated Skin and Skin Structure Infections | All Other Indications | ||
|---|---|---|---|---|
| ZYVOX 400 mg by mouth every 12 hours (n=548) | Clarithromycin 250 mg by mouth every 12 hours (n=537) | ZYVOX 600 mg every 12 hours (n=1498) | All Other Comparators* (n=1464) | |
| ||||
Headache | 8.8 | 8.4 | 5.7 | 4.4 |
Diarrhea | 8.2 | 6.1 | 8.3 | 6.4 |
Nausea | 5.1 | 4.5 | 6.6 | 4.6 |
Vomiting | 2.0 | 1.5 | 4.3 | 2.3 |
Dizziness | 2.6 | 3.0 | 1.8 | 1.5 |
Rash | 1.1 | 1.1 | 2.3 | 2.6 |
Anemia | 0.4 | 0 | 2.1 | 1.4 |
Taste alteration | 1.8 | 2.0 | 1.0 | 0.3 |
Vaginal moniliasis | 1.8 | 1.3 | 1.1 | 0.5 |
Oral moniliasis | 0.5 | 0 | 1.7 | 1.0 |
Abnormal liver function tests | 0.4 | 0.2 | 1.6 | 0.8 |
Fungal infection | 1.5 | 0.2 | 0.3 | 0.2 |
Tongue discoloration | 1.3 | 0 | 0.3 | 0 |
Localized abdominal pain | 1.3 | 0.6 | 1.2 | 0.8 |
Generalized abdominal pain | 0.9 | 0.4 | 1.2 | 1.0 |
Of the patients treated for uSSSIs, 3.5% of ZYVOX-treated and 2.4% of comparator-treated patients discontinued treatment due to drug-related adverse events. For all other indications, discontinuations due to drug-related adverse events occurred in 2.1% of ZYVOX-treated and 1.7% of comparator-treated patients. The most common reported drug-related adverse events leading to discontinuation of treatment were nausea, headache, diarrhea, and vomiting.
Pediatric Patients
The safety of ZYVOX formulations was evaluated in 215 pediatric patients ranging in age from birth through 11 years, and in 248 pediatric patients aged 5 through 17 years (146 of these 248 were age 5 through 11 and 102 were age 12 to 17). These patients were enrolled in two Phase 3 comparator-controlled clinical trials and were treated for up to 28 days. In the study of hospitalized pediatric patients (birth through 11 years) with Gram-positive infections, who were randomized 2 to 1 (linezolid: vancomycin), mortality was 6.0% (13/215) in the linezolid arm and 3.0% (3/101) in the vancomycin arm. However, given the severe underlying illness in the patient population, no causality could be established.
Of the pediatric patients treated for uSSSIs, 19.2% of ZYVOX-treated and 14.1% of comparator-treated patients experienced at least one drug-related adverse event. For all other indications, 18.8% of ZYVOX-treated and 34.3% of comparator-treated patients experienced at least one drug-related adverse event.
Table 3 shows the incidence of all-causality, treatment-emergent adverse reactions reported in more than 1% of pediatric patients (and more than 1 patient) in either treatment group in the comparator-controlled Phase 3 trials.
| ADVERSE REACTIONS | Uncomplicated Skin and Skin Structure Infections* | All Other Indications† | ||
|---|---|---|---|---|
| ZYVOX (n=248) | Cefadroxil (n=251) | ZYVOX (n=215) | Vancomycin (n=101) | |
| ||||
Diarrhea | 7.8 | 8.0 | 10.8 | 12.1 |
Vomiting | 2.9 | 6.4 | 9.4 | 9.1 |
Headache | 6.5 | 4.0 | 0.9 | 0 |
Anemia | 0 | 0 | 5.6 | 7.1 |
Thrombocytopenia | 0 | 0 | 4.7 | 2.0 |
Nausea | 3.7 | 3.2 | 1.9 | 0 |
Generalized abdominal pain | 2.4 | 2.8 | 0.9 | 2.0 |
Localized abdominal pain | 2.4 | 2.8 | 0.5 | 1.0 |
Loose stools | 1.6 | 0.8 | 2.3 | 3.0 |
Eosinophilia | 0.4 | 0.8 | 1.9 | 1.0 |
Pruritus at non-application site | 0.8 | 0.4 | 1.4 | 2.0 |
Vertigo | 1.2 | 0.4 | 0 | 0 |
Of the pediatric patients treated for uSSSIs, 1.6% of ZYVOX-treated and 2.4% of comparator-treated patients discontinued treatment due to drug-related adverse events. For all other indications, discontinuations due to drug-related adverse events occurred in 0.9% of ZYVOX-treated and 6.1% of comparator-treated patients.
Laboratory Abnormalities
ZYVOX has been associated with thrombocytopenia when used in doses up to and including 600 mg every 12 hours for up to 28 days. In Phase 3 comparator-controlled trials, the percentage of adult patients who developed a substantially low platelet count (defined as less than 75% of lower limit of normal and/or baseline) was 2.4% (range among studies: 0.3 to 10.0%) with ZYVOX and 1.5% (range among studies: 0.4 to 7.0%) with a comparator. In a study of hospitalized pediatric patients ranging in age from birth through 11 years, the percentage of patients who developed a substantially low platelet count (defined as less than 75% of lower limit of normal and/or baseline) was 12.9% with ZYVOX and 13.4% with vancomycin. In an outpatient study of pediatric patients aged from 5 through 17 years, the percentage of patients who developed a substantially low platelet count was 0% with ZYVOX and 0.4% with cefadroxil. Thrombocytopenia associated with the use of ZYVOX appears to be dependent on duration of therapy (generally greater than 2 weeks of treatment). The platelet counts for most patients returned to the normal range/baseline during the follow-up period. No related clinical adverse events were identified in Phase 3 clinical trials in patients developing thrombocytopenia. Bleeding events were identified in thrombocytopenic patients in a compassionate use program for ZYVOX; the role of linezolid in these events cannot be determined [see Warnings and Precautions (5.1)].
Changes seen in other laboratory parameters, without regard to drug relationship, revealed no substantial differences between ZYVOX and the comparators. These changes were generally not clinically significant, did not lead to discontinuation of therapy, and were reversible. The incidence of adult and pediatric patients with at least one substantially abnormal hematologic or serum chemistry value is presented in Tables 4, 5, 6, and 7.
| Laboratory Assay | Uncomplicated Skin and Skin Structure Infections | All Other Indications | ||
|---|---|---|---|---|
| ZYVOX 400 mg every 12 hours | Clarithromycin 250 mg every 12 hours | ZYVOX 600 mg every 12 hours | All Other Comparators† | |
| ||||
Hemoglobin (g/dL) | 0.9 | 0.0 | 7.1 | 6.6 |
Platelet count (× 103/mm3) | 0.7 | 0.8 | 3.0 | 1.8 |
WBC (× 103/mm3) | 0.2 | 0.6 | 2.2 | 1.3 |
Neutrophils (× 103/mm3) | 0.0 | 0.2 | 1.1 | 1.2 |
| Laboratory Assay | Uncomplicated Skin and Skin Structure Infections | All Other Indications | ||
|---|---|---|---|---|
| ZYVOX 400 mg every 12 hours | Clarithromycin 250 mg every 12 hours | ZYVOX 600 mg every 12 hours | All Other Comparators† | |
| ||||
AST (U/L) | 1.7 | 1.3 | 5.0 | 6.8 |
ALT (U/L) | 1.7 | 1.7 | 9.6 | 9.3 |
LDH (U/L) | 0.2 | 0.2 | 1.8 | 1.5 |
Alkaline phosphatase (U/L) | 0.2 | 0.2 | 3.5 | 3.1 |
Lipase (U/L) | 2.8 | 2.6 | 4.3 | 4.2 |
Amylase (U/L) | 0.2 | 0.2 | 2.4 | 2.0 |
Total bilirubin (mg/dL) | 0.2 | 0.0 | 0.9 | 1.1 |
BUN (mg/dL) | 0.2 | 0.0 | 2.1 | 1.5 |
Creatinine (mg/dL) | 0.2 | 0.0 | 0.2 | 0.6 |
| Laboratory Assay | Uncomplicated Skin and Skin Structure Infections† | All Other Indications‡ | ||
|---|---|---|---|---|
| ZYVOX | Cefadroxil | ZYVOX | Vancomycin | |
| ||||
Hemoglobin (g/dL) | 0.0 | 0.0 | 15.7 | 12.4 |
Platelet count (× 103/mm3) | 0.0 | 0.4 | 12.9 | 13.4 |
WBC (× 103/mm3) | 0.8 | 0.8 | 12.4 | 10.3 |
Neutrophils (× 103/mm3) | 1.2 | 0.8 | 5.9 | 4.3 |
| Laboratory Assay | Uncomplicated Skin and Skin Structure Infections† | All Other Indications‡ | ||
|---|---|---|---|---|
| ZYVOX | Cefadroxil | ZYVOX | Vancomycin | |
| ||||
ALT (U/L) | 0.0 | 0.0 | 10.1 | 12.5 |
Lipase (U/L) | 0.4 | 1.2 | --- | --- |
Amylase (U/L) | --- | --- | 0.6 | 1.3 |
Total bilirubin (mg/dL) | --- | --- | 6.3 | 5.2 |
Creatinine (mg/dL) | 0.4 | 0.0 | 2.4 | 1.0 |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ZYVOX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
- •
- Anaphylaxis, angioedema, bullous skin disorders including severe cutaneous adverse reactions (SCAR) such as toxic epidermal necrolysis and Stevens-Johnson syndrome, and hypersensitivity vasculitis.
- •
- Myelosuppression (including anemia, leukopenia, pancytopenia, and thrombocytopenia). Thrombocytopenia has been reported more often in patients with severe renal impairment and in patients with moderate to severe hepatic impairment [see Warnings and Precautions (5.1)]; sideroblastic anemia.
- •
- Peripheral neuropathy, and optic neuropathy sometimes progressing to loss of vision [see Warnings and Precautions (5.2)].
- •
- Serotonin syndrome has been reported in patients receiving concomitant serotonergic agents, including antidepressants such as selective serotonin reuptake inhibitors (SSRIs) and opioids, and ZYVOX [see Warnings and Precautions (5.3)].
- •
- Lactic acidosis [see Warnings and Precautions (5.7)]. Although these reports have primarily been in patients treated for longer than the maximum recommended duration of 28 days, these events have also been reported in patients receiving shorter courses of therapy.
- •
- Convulsions [see Warnings and Precautions (5.8)].
- •
- Rhabdomyolysis [see Warnings and Precautions (5.9)].
- •
- Hypoglycemia, including symptomatic episodes [see Warnings and Precautions (5.10)].
- •
- Hyponatremia and/or Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH) [see Warnings and Precautions (5.11)].
- •
- Superficial tooth discoloration and tongue discoloration have been reported with the use of linezolid. The tooth discoloration was removable with professional dental cleaning (manual descaling) in cases with known outcome.
Drug Interactions
7 DRUG INTERACTIONS
7.1 Monoamine Oxidase Inhibitors
Linezolid is a reversible, nonselective inhibitor of monoamine oxidase [see Contraindications (4.2) and Clinical Pharmacology (12.3)].
7.2 Adrenergic and Serotonergic Agents
Linezolid has the potential for interaction with adrenergic and serotonergic agents [see Warnings and Precautions (5.3, 5.6) and Clinical Pharmacology (12.3)].
Use in Specific Populations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from published and postmarketing case reports with linezolid use in pregnant women have not identified a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. When administered during organogenesis, linezolid did not cause malformations in mice, rats, or rabbits at maternal exposure levels approximately 6.5 times (mice), equivalent to (rats), or 0.06 times (rabbits) the clinical therapeutic exposure, based on AUCs. However, embryo-fetal lethality was observed in mice at 6.5 times the estimated human exposure. When female rats were dosed during organogenesis through lactation, postnatal survival of pups was decreased at doses approximately equivalent to the estimated human exposure based on AUCs (see Data).
The background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Animal Data
In mice, embryo-fetal toxicities were observed only at doses that caused maternal toxicity (clinical signs and reduced body weight gain). An oral dose of 450 mg/kg/day given from Gestation Day (GD) 6–16 (6.5 times the estimated human exposure based on AUCs) correlated with increased postimplantational embryo death, including total litter loss, decreased fetal body weights, and an increased incidence of costal cartilage fusion. Neither maternal nor embryo-fetal toxicities were observed at doses up to 150 mg/kg/day. Fetal malformations were not observed.
In rats, fetal toxicity was observed at 15 and 50 mg/kg/day administered orally from GD 6–17 (exposures 0.22 times to approximately equivalent to the estimated human exposure, respectively, based on AUCs). The effects consisted of decreased fetal body weights and reduced ossification of sternebrae, a finding often seen in association with decreased fetal body weights. Fetal malformations were not observed. Maternal toxicity, in the form of reduced body weight gain, was seen at 50 mg/kg/day.
In rabbits, reduced fetal body weight occurred only in the presence of maternal toxicity (clinical signs, reduced body weight gain and food consumption) when administered at an oral dose of 15 mg/kg/day given from GD 6–20 (0.06 times the estimated human exposure based on AUCs). Fetal malformations were not observed.
When female rats were treated with 50 mg/kg/day (approximately equivalent to the estimated human exposure based on AUCs) of linezolid during pregnancy and lactation (GD 6 through Lactation Day 20), survival of pups was decreased on postnatal days 1 to 4. Male and female pups permitted to mature to reproductive age, when mated, showed an increase in preimplantation loss.
8.2 Lactation
Risk Summary
Linezolid is present in breast milk. Based on data from available published case reports, the daily dose of linezolid that the infant would receive from breastmilk would be approximately 6% to 9% of the recommended therapeutic infant dose (10 mg/kg every 8 hours). There is no information on the effects of linezolid on the breastfed infant; however, diarrhea and vomiting were the most common adverse reactions reported in clinical trials in infants receiving linezolid therapeutically [see Adverse Reactions (6.1)] and (see Clinical Considerations). There is no information on the effects of linezolid on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for linezolid and any potential adverse effects on the breastfed child from linezolid or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Infertility
Males
Based on findings from studies in rats, ZYVOX may reversibly impair fertility in male patients [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of ZYVOX for the treatment of pediatric patients with the following infections are supported by evidence from adequate and well-controlled studies in adults, pharmacokinetic data in pediatric patients, and additional data from a comparator-controlled study of Gram-positive infections in pediatric patients ranging in age from birth through 11 years [see Indications and Usage (1), Clinical Pharmacology (12.3) and Clinical Studies (14)]:
- •
- nosocomial pneumonia
- •
- complicated skin and skin structure infections
- •
- community-acquired pneumonia (also supported by evidence from an uncontrolled study in patients ranging in age from 8 months through 12 years)
- •
- vancomycin-resistant Enterococcus faecium infections
The safety and effectiveness of ZYVOX for the treatment of pediatric patients with the following infection have been established in a comparator-controlled study in pediatric patients ranging in age from 5 through 17 years [see Clinical Studies (14)]:
- •
- uncomplicated skin and skin structure infections caused by Staphylococcus aureus (methicillin-susceptible strains only) or Streptococcus pyogenes
Pharmacokinetic information generated in pediatric patients with ventriculoperitoneal shunts showed variable cerebrospinal fluid (CSF) linezolid concentrations following single and multiple dosing of linezolid; therapeutic concentrations were not consistently achieved or maintained in the CSF. Therefore, the use of linezolid for the empiric treatment of pediatric patients with central nervous system infections is not recommended.
The pharmacokinetics of linezolid have been evaluated in pediatric patients from birth to 17 years of age. In general, weight-based clearance of linezolid gradually decreases with increasing age of pediatric patients. However, in preterm (gestational age < 34 weeks) neonates < 7 days of age, linezolid clearance is often lower than in full-term neonates < 7 days of age. Consequently, preterm neonates < 7 days of age may need an alternative linezolid dosing regimen of 10 mg/kg every 12 hours [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)].
In limited clinical experience, 5 out of 6 (83%) pediatric patients with infections due to Gram-positive pathogens with minimum inhibitory concentrations (MICs) of 4 mcg/mL treated with ZYVOX had clinical cures. However, pediatric patients exhibit wider variability in linezolid clearance and systemic exposure (AUC) compared with adults. In pediatric patients with a sub-optimal clinical response, particularly those with pathogens with MIC of 4 mcg/mL, lower systemic exposure, site and severity of infection, and the underlying medical condition should be considered when assessing clinical response [see Clinical Pharmacology (12.3) and Dosage and Administration (2)].
8.5 Geriatric Use
Of the 2,046 patients treated with ZYVOX in Phase 3 comparator-controlled clinical trials, 589 (29%) were 65 years or older and 253 (12%) were 75 years or older. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Overdosage
10 OVERDOSAGE
In the event of overdosage, supportive care is advised, with maintenance of glomerular filtration. Hemodialysis may facilitate more rapid elimination of linezolid. In a Phase 1 clinical trial, approximately 30% of a dose of linezolid was removed during a 3-hour hemodialysis session beginning 3 hours after the dose of linezolid was administered. Data are not available for removal of linezolid with peritoneal dialysis or hemoperfusion. Clinical signs of acute toxicity in animals were decreased activity and ataxia in rats and vomiting and tremors in dogs treated with 3,000 mg/kg/day and 2,000 mg/kg/day, respectively.
Description
11 DESCRIPTION
ZYVOX I.V. Injection, ZYVOX Tablets, and ZYVOX for Oral Suspension contain linezolid, which is a synthetic antibacterial agent of the oxazolidinone class. The chemical name for linezolid is (S)-N-[[3-[3-Fluoro-4-(4-morpholinyl)phenyl]-2-oxo-5-oxazolidinyl] methyl]-acetamide.
The empirical formula is C16H20FN3O4. Its molecular weight is 337.35, and its chemical structure is represented below:
ZYVOX I.V. Injection is supplied as a ready-to-use sterile isotonic solution for intravenous infusion. Each mL contains 2 mg of linezolid. Inactive ingredients are dextrose monohydrate 50.24 mg/mL in an aqueous vehicle for intravenous administration, sodium citrate dihydrate 1.64 mg/mL, and citric acid anhydrous 0.85 mg/mL. Sodium hydroxide NF and/or hydrochloric acid NF are used to adjust the pH. The sodium (Na+) content is 0.38 mg/mL (5 mEq/300 mL bag and 1.7 mEq/100 mL bag).
ZYVOX Tablet for oral administration contains 600 mg linezolid as a film-coated compressed tablet. Inactive ingredients are carnauba wax, corn starch, hydroxypropylcellulose, hypromellose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, sodium starch glycolate, and titanium dioxide. The sodium (Na+) content is 2.92 mg per 600 mg tablet (0.1 mEq/tablet).
ZYVOX for Oral Suspension is supplied as an orange-flavored granule/powder for constitution into a suspension for oral administration. Following constitution, each 5 mL contains 100 mg of linezolid. Inactive ingredients are aspartame, citric acid, colloidal silicon dioxide, flavors, mannitol, microcrystalline cellulose and carboxymethylcellulose sodium, sodium benzoate, sodium chloride, sodium citrate, sucrose, and xanthan gum [see Patient Counseling Information (17)]. The sodium (Na+) content is 8.52 mg/5 mL (0.4 mEq/5 mL).
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
In a randomized, positive- and placebo-controlled crossover thorough QT study, 40 healthy subjects were administered a single ZYVOX 600 mg dose via a 1 hour IV infusion, a single ZYVOX 1,200 mg dose via a 1 hour IV infusion, placebo, and a single oral dose of positive control. At both the 600 mg and 1,200 mg ZYVOX doses, no significant effect on QTc interval was detected at peak plasma concentration or at any other time.
12.3 Pharmacokinetics
The mean pharmacokinetic parameters of linezolid in adults after single and multiple oral and intravenous doses are summarized in Table 8. Plasma concentrations of linezolid at steady-state after oral doses of 600 mg given every 12 hours are shown in Figure 1.
| Dose of Linezolid | Cmax mcg/mL | Cmin mcg/mL | Tmax hrs | AUC * mcg∙h/mL | t1/2 hrs | CL mL/min |
|---|---|---|---|---|---|---|
| Cmax = Maximum plasma concentration; Cmin = Minimum plasma concentration; Tmax = Time to Cmax; AUC = Area under concentration-time curve; t1/2 = Elimination half-life; CL = Systemic clearance | ||||||
400 mg tablet | ||||||
single dose † | 8.10 | --- | 1.52 | 55.10 | 5.20 | 146 |
every 12 hours | 11.00 | 3.08 | 1.12 | 73.40 | 4.69 | 110 |
600 mg tablet | ||||||
single dose | 12.70 | --- | 1.28 | 91.40 | 4.26 | 127 |
every 12 hours | 21.20 | 6.15 | 1.03 | 138.00 | 5.40 | 80 |
600 mg IV injection ‡ | ||||||
single dose | 12.90 | --- | 0.50 | 80.20 | 4.40 | 138 |
every 12 hours | 15.10 | 3.68 | 0.51 | 89.70 | 4.80 | 123 |
600 mg oral suspension | ||||||
single dose | 11.00 | --- | 0.97 | 80.80 | 4.60 | 141 |
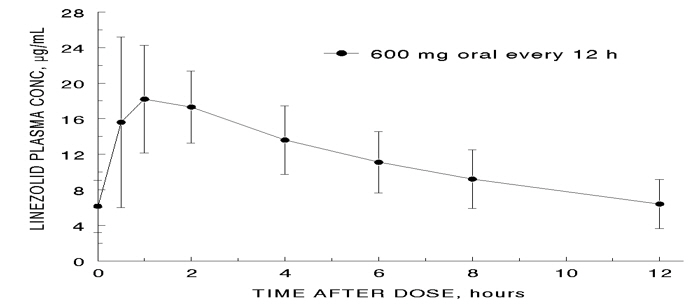
Figure 1. Plasma Concentrations of Linezolid in Adults at Steady-State Following Oral Dosing Every 12 Hours (Mean ± Standard Deviation, n=16)
Absorption
Linezolid is extensively absorbed after oral dosing. Maximum plasma concentrations are reached approximately 1 to 2 hours after dosing, and the absolute bioavailability is approximately 100%. Therefore, linezolid may be given orally or intravenously without dose adjustment.
Linezolid may be administered without regard to the timing of meals. The time to reach the maximum concentration is delayed from 1.5 hours to 2.2 hours and Cmax is decreased by about 17% when high fat food is given with linezolid. However, the total exposure measured as AUC0–∞ is similar under both conditions.
Distribution
Animal and human pharmacokinetic studies have demonstrated that linezolid readily distributes to well-perfused tissues. The plasma protein binding of linezolid is approximately 31% and is concentration-independent. The volume of distribution of linezolid at steady-state averaged 40 to 50 liters in healthy adult volunteers.
Linezolid concentrations have been determined in various fluids from a limited number of subjects in Phase 1 volunteer studies following multiple dosing of linezolid. The ratio of linezolid in saliva relative to plasma was 1.2 to 1 and the ratio of linezolid in sweat relative to plasma was 0.55 to 1.
Metabolism
Linezolid is primarily metabolized by oxidation of the morpholine ring, which results in two inactive ring-opened carboxylic acid metabolites: the aminoethoxyacetic acid metabolite (A), and the hydroxyethyl glycine metabolite (B). Formation of metabolite A is presumed to be formed via an enzymatic pathway whereas metabolite B is mediated by a non-enzymatic chemical oxidation mechanism in vitro. In vitro studies have demonstrated that linezolid is minimally metabolized and may be mediated by human cytochrome P450. However, the metabolic pathway of linezolid is not fully understood.
Excretion
Nonrenal clearance accounts for approximately 65% of the total clearance of linezolid. Under steady-state conditions, approximately 30% of the dose appears in the urine as linezolid, 40% as metabolite B, and 10% as metabolite A. The mean renal clearance of linezolid is 40 mL/min which suggests net tubular reabsorption. Virtually no linezolid appears in the feces, while approximately 6% of the dose appears in the feces as metabolite B, and 3% as metabolite A.
A small degree of nonlinearity in clearance was observed with increasing doses of linezolid, which appears to be due to lower renal and nonrenal clearance of linezolid at higher concentrations. However, the difference in clearance was small and was not reflected in the apparent elimination half-life.
Specific Populations
Geriatric Patients
The pharmacokinetics of linezolid are not significantly altered in elderly patients (65 years or older). Therefore, dose adjustment for geriatric patients is not necessary.
Pediatric Patients
The pharmacokinetics of linezolid following a single intravenous dose were investigated in pediatric patients ranging in age from birth through 17 years (including premature and full-term neonates), in healthy adolescent subjects ranging in age from 12 through 17 years, and in pediatric patients ranging in age from 1 week through 12 years. The pharmacokinetic parameters of linezolid are summarized in Table 9 for the pediatric populations studied and healthy adult subjects after administration of single intravenous doses.
The Cmax and the volume of distribution (Vss) of linezolid are similar regardless of age in pediatric patients. However, plasma clearance of linezolid varies as a function of age. With the exclusion of pre-term neonates less than one week of age, weight-based clearance is most rapid in the youngest age groups ranging from < 1 week old to 11 years, resulting in lower single-dose systemic exposure (AUC) and a shorter half-life as compared with adults. As the age of pediatric patients increases, the weight-based clearance of linezolid gradually decreases, and by adolescence mean clearance values approach those observed for the adult population. There is increased inter-subject variability in linezolid clearance and systemic drug exposure (AUC) across all pediatric age groups as compared with adults.
Similar mean daily AUC values were observed in pediatric patients from birth to 11 years of age dosed every 8 hours relative to adolescents or adults dosed every 12 hours. Therefore, the dosage for pediatric patients up to 11 years of age should be 10 mg/kg every 8 hours. Pediatric patients 12 years and older should receive 600 mg every 12 hours [see Dosage and Administration (2)].
| Age Group | Cmax mcg/mL | Vss L/kg | AUC* mcg∙h/mL | t 1/2 hrs | CL mL/min/kg |
|---|---|---|---|---|---|
| Cmax = Maximum plasma concentration; Vss= Volume of distribution; AUC = Area under concentration-time curve; t1/2 = Apparent elimination half-life; CL = Systemic clearance normalized for body weight | |||||
| |||||
Neonatal Patients | |||||
Pre-term† | 12.7 (30%) | 0.81 (24%) | 108 (47%) | 5.6 (46%) | 2.0 (52%) |
< 1 week (N=9)‡ | [9.6, 22.2] | [0.43, 1.05] | [41, 191] | [2.4, 9.8] | [0.9, 4.0] |
Full-term§ | 11.5 (24%) | 0.78 (20%) | 55 (47%) | 3.0 (55%) | 3.8 (55%) |
< 1 week (N=10)‡ | [8.0, 18.3] | [0.45, 0.96] | [19, 103] | [1.3, 6.1] | [1.5, 8.8] |
Full-term§ | 12.9 (28%) | 0.66 (29%) | 34 (21%) | 1.5 (17%) | 5.1 (22%) |
≥ 1 week to ≤ 28 days (N=10)‡ | [7.7, 21.6] | [0.35, 1.06] | [23, 50] | [1.2, 1.9] | [3.3, 7.2] |
Infant Patients | |||||
> 28 days to < 3 Months (N=12)‡ | 11.0 (27%) | 0.79 (26%) | 33 (26%) | 1.8 (28%) | 5.4 (32%) |
Pediatric Patients | |||||
3 months through 11 years‡ (N=59) | 15.1 (30%) | 0.69 (28%) | 58 (54%) | 2.9 (53%) | 3.8 (53%) |
Adolescent Subjects and Patients | |||||
12 through 17 years¶ | 16.7 (24%) | 0.61 (15%) | 95 (44%) | 4.1 (46%) | 2.1 (53%) |
Adult Subjects# | 12.5 (21%) | 0.65 (16%) | 91 (33%) | 4.9 (35%) | 1.7 (34%) |
(N= 29) | [8.2, 19.3] | [0.45, 0.84] | [53, 155] | [1.8, 8.3] | [0.9, 3.3] |
Gender
Females have a slightly lower volume of distribution of linezolid than males. Plasma concentrations are higher in females than in males, which is partly due to body weight differences. After a 600-mg dose, mean oral clearance is approximately 38% lower in females than in males. However, there are no significant gender differences in mean apparent elimination-rate constant or half-life. Thus, drug exposure in females is not expected to substantially increase beyond levels known to be well tolerated. Therefore, dose adjustment by gender does not appear to be necessary.
Renal Impairment
The pharmacokinetics of the parent drug, linezolid, are not altered in patients with any degree of renal impairment; however, the two primary metabolites of linezolid accumulate in patients with renal impairment, with the amount of accumulation increasing with the severity of renal dysfunction (see Table 10). The pharmacokinetics of linezolid and its two metabolites have also been studied in patients with end-stage renal disease (ESRD) receiving hemodialysis. In the ESRD study, 14 patients were dosed with linezolid 600 mg every 12 hours for 14.5 days (see Table 11). Because similar plasma concentrations of linezolid are achieved regardless of renal function, no dose adjustment is recommended for patients with renal impairment. However, given the absence of information on the clinical significance of accumulation of the primary metabolites, use of linezolid in patients with renal impairment should be weighed against the potential risks of accumulation of these metabolites. Both linezolid and the two metabolites are eliminated by hemodialysis. No information is available on the effect of peritoneal dialysis on the pharmacokinetics of linezolid. Approximately 30% of a dose was eliminated in a 3-hour hemodialysis session beginning 3 hours after the dose of linezolid was administered; therefore, linezolid should be given after hemodialysis.
| Parameter | Healthy Subjects CLCR > 80 mL/min | Moderate Renal Impairment 30 < CLCR < 80 mL/min | Severe Renal Impairment 10 < CLCR < 30 mL/min |
|---|---|---|---|
| |||
LINEZOLID | |||
AUC0–∞, mcg h/mL | 110 (22) | 128 (53) | 127 (66) |
t1/2, hours | 6.4 (2.2) | 6.1 (1.7) | 7.1 (3.7) |
METABOLITE A | |||
AUC0–48, mcg h/mL | 7.6 (1.9) | 11.7 (4.3) | 56.5 (30.6) |
t1/2, hours | 6.3 (2.1) | 6.6 (2.3) | 9.0 (4.6) |
METABOLITE B* | |||
AUC0–48, mcg h/mL | 30.5 (6.2) | 51.1 (38.5) | 203 (92) |
t1/2, hours | 6.6 (2.7) | 9.9 (7.4) | 11.0 (3.9) |
| Parameter | ESRD Subjects* |
|---|---|
LINEZOLID | |
AUC0–12, mcg h/mL (after last dose) | 181 (52.3) |
t1/2, h (after last dose) | 8.3 (2.4) |
METABOLITE A | |
AUC0–12, mcg h/mL (after last dose) | 153 (40.6) |
t1/2, h (after last dose) | 15.9 (8.5) |
METABOLITE B† | |
AUC0–12, mcg h/mL (after last dose) | 356 (99.7) |
t1/2, h (after last dose) | 34.8 (23.1) |
Hepatic Impairment
The pharmacokinetics of linezolid are not altered in patients (n=7) with mild-to-moderate hepatic impairment (Child-Pugh class A or B). On the basis of the available information, no dose adjustment is recommended for patients with mild-to-moderate hepatic impairment. The pharmacokinetics of linezolid in patients with severe hepatic impairment have not been evaluated.
Drug Interactions
Drugs Metabolized by Cytochrome P450
Linezolid is not an inducer of cytochrome P450 (CYP450) in rats. In addition, linezolid does not inhibit the activities of clinically significant human CYP isoforms (e.g., 1A2, 2C9, 2C19, 2D6, 2E1, 3A4). Therefore, linezolid is not expected to affect the pharmacokinetics of other drugs metabolized by these major enzymes. Concurrent administration of linezolid does not substantially alter the pharmacokinetic characteristics of (S)-warfarin, which is extensively metabolized by CYP2C9. Drugs such as warfarin and phenytoin, which are CYP2C9 substrates, may be given with linezolid without changes in dosage regimen.
Antibacterial Drugs
Antioxidants
The potential for drug-drug interactions with linezolid and the antioxidants Vitamin C and Vitamin E was studied in healthy volunteers. Subjects were administered a 600 mg oral dose of linezolid on Day 1, and another 600 mg dose of linezolid on Day 8. On Days 2–9, subjects were given either Vitamin C (1,000 mg/day) or Vitamin E (800 IU/ day). The AUC0–∞ of linezolid increased 2.3% when co-administered with Vitamin C and 10.9% when co-administered with Vitamin E. No linezolid dose adjustment is recommended during co-administration with Vitamin C or Vitamin E.
Strong CYP 3A4 Inducers
Rifampin: The effect of rifampin on the pharmacokinetics of linezolid was evaluated in a study of 16 healthy adult males. Volunteers were administered oral linezolid 600 mg twice daily for 5 doses with and without rifampin 600 mg once daily for 8 days. Co-administration of rifampin with linezolid resulted in a 21% decrease in linezolid Cmax [90% CI, 15% – 27%] and a 32% decrease in linezolid AUC0–12 [90% CI, 27% – 37%]. The clinical significance of this interaction is unknown. The mechanism of this interaction is not fully understood and may be related to the induction of hepatic enzymes. Other strong inducers of hepatic enzymes (e.g. carbamazepine, phenytoin, phenobarbital) could cause a similar or smaller decrease in linezolid exposure.
Monoamine Oxidase Inhibition
Linezolid is a reversible, nonselective inhibitor of monoamine oxidase. Therefore, linezolid has the potential for interaction with adrenergic and serotonergic agents.
Adrenergic Agents
Some individuals receiving ZYVOX may experience a reversible enhancement of the pressor response to indirect-acting sympathomimetic agents, vasopressor or dopaminergic agents. Commonly used drugs such as phenylpropanolamine and pseudoephedrine have been specifically studied. Initial doses of adrenergic agents, such as dopamine or epinephrine, should be reduced and titrated to achieve the desired response.
Tyramine: A significant pressor response has been observed in normal adult subjects receiving linezolid and tyramine doses of more than 100 mg. Therefore, patients receiving linezolid need to avoid consuming large amounts of foods or beverages with high tyramine content [see Patient Counseling Information (17)].
Pseudoephedrine HCl or phenylpropanolamine HCl: A reversible enhancement of the pressor response of either pseudoephedrine HCl (PSE) or phenylpropanolamine HCl (PPA) is observed when linezolid is administered to healthy normotensive subjects [see Warnings and Precautions (5.6) and Drug Interactions (7)]. A similar study has not been conducted in hypertensive patients. The interaction studies conducted in normotensive subjects evaluated the blood pressure and heart rate effects of placebo, PPA or PSE alone, linezolid alone, and the combination of steady-state linezolid (600 mg every 12 hours for 3 days) with two doses of PPA (25 mg) or PSE (60 mg) given 4 hours apart. Heart rate was not affected by any of the treatments. Blood pressure was increased with both combination treatments. Maximum blood pressure levels were seen 2 to 3 hours after the second dose of PPA or PSE, and returned to baseline 2 to 3 hours after peak. The results of the PPA study follow, showing the mean (and range) maximum systolic blood pressure in mm Hg: placebo = 121 (103 to 158); linezolid alone = 120 (107 to 135); PPA alone = 125 (106 to 139); PPA with linezolid = 147 (129 to 176). The results from the PSE study were similar to those in the PPA study. The mean maximum increase in systolic blood pressure over baseline was 32 mm Hg (range: 20–52 mm Hg) and 38 mm Hg (range: 18–79 mm Hg) during co-administration of linezolid with pseudoephedrine or phenylpropanolamine, respectively.
Serotonergic Agents
Dextromethorphan: The potential drug-drug interaction with dextromethorphan was studied in healthy volunteers. Subjects were administered dextromethorphan (two 20-mg doses given 4 hours apart) with or without linezolid. No serotonin syndrome effects (confusion, delirium, restlessness, tremors, blushing, diaphoresis, hyperpyrexia) have been observed in normal subjects receiving linezolid and dextromethorphan.
12.4 Microbiology
Mechanism of Action
Linezolid is a synthetic antibacterial agent of the oxazolidinone class, which has clinical utility in the treatment of infections caused by aerobic Gram-positive bacteria. The in vitro spectrum of activity of linezolid also includes certain Gram-negative bacteria and anaerobic bacteria. Linezolid binds to a site on the bacterial 23S ribosomal RNA of the 50S subunit and prevents the formation of a functional 70S initiation complex, which is essential for bacterial reproduction. The results of time-kill studies have shown linezolid to be bacteriostatic against enterococci and staphylococci. For streptococci, linezolid was found to be bactericidal for the majority of isolates.
Resistance
In vitro studies have shown that point mutations in the 23S rRNA are associated with linezolid resistance. Reports of vancomycin-resistant Enterococcus faecium becoming resistant to linezolid during its clinical use have been published. There are reports of Staphylococcus aureus (methicillin-resistant) developing resistance to linezolid during clinical use. The linezolid resistance in these organisms is associated with a point mutation in the 23S rRNA (substitution of thymine for guanine at position 2576) of the organism. Organisms resistant to oxazolidinones via mutations in chromosomal genes encoding 23S rRNA or ribosomal proteins (L3 and L4) are generally cross-resistant to linezolid. Also linezolid resistance in staphylococci mediated by the enzyme methyltransferase has been reported. This resistance is mediated by the cfr (chloramphenicol-florfenicol) gene located on a plasmid which is transferable between staphylococci.
Interaction with Other Antimicrobial Drugs
In vitro studies have demonstrated additivity or indifference between linezolid and vancomycin, gentamicin, rifampin, imipenem-cilastatin, aztreonam, ampicillin, or streptomycin.
Linezolid has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1)].
Gram-positive bacteria
Enterococcus faecium (vancomycin-resistant isolates only)
Staphylococcus aureus (including methicillin-resistant isolates)
Streptococcus agalactiae
Streptococcus pneumoniae
Streptococcus pyogenes
The following in vitro data are available, but their clinical significance is unknown. Greater than 90% of the following bacteria exhibit an in vitro MIC less than or equal to the linezolid-susceptible breakpoint for organisms of similar genus. The safety and effectiveness of linezolid in treating clinical infections due to these bacteria have not been established in adequate and well-controlled clinical trials.
Gram-positive bacteria
Enterococcus faecalis (including vancomycin-resistant isolates)
Enterococcus faecium (vancomycin-susceptible isolates)
Staphylococcus epidermidis (including methicillin-resistant isolates)
Staphylococcus haemolyticus
Viridans group streptococci
Gram-negative bacteria
Pasteurella multocida
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
Nonclinical Toxicology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Lifetime studies in animals have not been conducted to evaluate the carcinogenic potential of linezolid. Neither mutagenic nor clastogenic potential was found in a battery of tests including: assays for mutagenicity (Ames bacterial reversion and CHO cell mutation), an in vitro unscheduled DNA synthesis (UDS) assay, an in vitro chromosome aberration assay in human lymphocytes, and an in vivo mouse micronucleus assay.
Linezolid did not affect the fertility or reproductive performance of adult female rats given oral doses of up to 100 mg/kg/day for 14 days prior to mating through Gestation Day 7. It reversibly decreased fertility and reproductive performance in adult male rats when given at doses ≥ 50 mg/kg/day, with exposures approximately equal to or greater than the expected human exposure level (exposure comparisons are based on AUCs). The reversible fertility effects were mediated through altered spermatogenesis. Affected spermatids contained abnormally formed and oriented mitochondria and were non-viable. Epithelial cell hypertrophy and hyperplasia in the epididymis was observed in conjunction with decreased fertility. Similar epididymal changes were not seen in dogs.
In sexually mature male rats exposed to drug as juveniles, mildly decreased fertility was observed following treatment with linezolid through most of their period of sexual development (50 mg/kg/day from days 7 to 36 of age, and 100 mg/kg/day from days 37 to 55 of age), with exposures up to 1.7 times greater than mean AUCs observed in pediatric patients aged 3 months to 11 years. Decreased fertility was not observed with shorter treatment periods, corresponding to exposure in utero through the early neonatal period (gestation day 6 through postnatal day 5), neonatal exposure (postnatal days 5 to 21), or to juvenile exposure (postnatal days 22 to 35). Reversible reductions in sperm motility and altered sperm morphology were observed in rats treated from postnatal day 22 to 35.
13.2 Animal Toxicology and/or Pharmacology
Target organs of linezolid toxicity were similar in juvenile and adult rats and dogs. Dose- and time-dependent myelosuppression, as evidenced by bone marrow hypocellularity/decreased hematopoiesis, decreased extramedullary hematopoiesis in spleen and liver, and decreased levels of circulating erythrocytes, leukocytes, and platelets have been seen in animal studies. Lymphoid depletion occurred in thymus, lymph nodes, and spleen. Generally, the lymphoid findings were associated with anorexia, weight loss, and suppression of body weight gain, which may have contributed to the observed effects.
In rats administered linezolid orally for 6 months, non-reversible, minimal to mild axonal degeneration of sciatic nerves was observed at 80 mg/kg/day; minimal degeneration of the sciatic nerve was also observed in 1 male at this dose level at a 3-month interim necropsy. Sensitive morphologic evaluation of perfusion-fixed tissues was conducted to investigate evidence of optic nerve degeneration. Minimal to moderate optic nerve degeneration was evident in 2 male rats after 6 months of dosing, but the direct relationship to drug was equivocal because of the acute nature of the finding and its asymmetrical distribution. The nerve degeneration observed was microscopically comparable to spontaneous unilateral optic nerve degeneration reported in aging rats and may be an exacerbation of common background change.
These effects were observed at exposure levels that are comparable to those observed in some human subjects. The hematopoietic and lymphoid effects were reversible, although in some studies, reversal was incomplete within the duration of the recovery period.
Clinical Studies
14 CLINICAL STUDIES
14.1 Adults
Nosocomial Pneumonia
Adult patients with clinically and radiologically documented nosocomial pneumonia were enrolled in a randomized, multi-center, double-blind trial. Patients were treated for 7 to 21 days. One group received ZYVOX I.V. Injection 600 mg every 12 hours, and the other group received vancomycin 1 g every 12 hours intravenously. Both groups received concomitant aztreonam (1 to 2 g every 8 hours intravenously), which could be continued if clinically indicated. There were 203 linezolid-treated and 193 vancomycin-treated patients enrolled in the study. One hundred twenty-two (60%) linezolid-treated patients and 103 (53%) vancomycin-treated patients were clinically evaluable. The cure rates in clinically evaluable patients were 57% for linezolid-treated patients and 60% for vancomycin-treated patients. The cure rates in clinically evaluable patients with ventilator-associated pneumonia were 47% for linezolid-treated patients and 40% for vancomycin-treated patients. A modified intent-to-treat (MITT) analysis of 94 linezolid-treated patients and 83 vancomycin-treated patients included subjects who had a pathogen isolated before treatment. The cure rates in the MITT analysis were 57% in linezolid-treated patients and 46% in vancomycin-treated patients. The cure rates by pathogen for microbiologically evaluable patients are presented in Table 12.
| Cured | ||
|---|---|---|
| Pathogen | ZYVOX n/N (%) | Vancomycin n/N (%) |
Staphylococcus aureus | 23/38 (61) | 14/23 (61) |
Methicillin-resistant S. aureus | 13/22 (59) | 7/10 (70) |
Streptococcus pneumoniae | 9/9 (100) | 9/10 (90) |
Complicated Skin and Skin Structure Infections
Adult patients with clinically documented complicated skin and skin structure infections were enrolled in a randomized, multi-center, double-blind, double-dummy trial comparing study medications administered intravenously followed by medications given orally for a total of 10 to 21 days of treatment. One group of patients received ZYVOX I.V. Injection 600 mg every 12 hours followed by ZYVOX Tablets 600 mg every 12 hours; the other group received oxacillin 2 g every 6 hours intravenously followed by dicloxacillin 500 mg every 6 hours orally. Patients could receive concomitant aztreonam if clinically indicated. There were 400 linezolid-treated and 419 oxacillin-treated patients enrolled in the study. Two hundred forty-five (61%) linezolid-treated patients and 242 (58%) oxacillin-treated patients were clinically evaluable. The cure rates in clinically evaluable patients were 90% in linezolid-treated patients and 85% in oxacillin-treated patients. A modified intent-to-treat (MITT) analysis of 316 linezolid-treated patients and 313 oxacillin-treated patients included subjects who met all criteria for study entry. The cure rates in the MITT analysis were 86% in linezolid-treated patients and 82% in oxacillin-treated patients. The cure rates by pathogen for microbiologically evaluable patients are presented in Table 13.
| Cured | ||
|---|---|---|
| Pathogen | ZYVOX n/N (%) | Oxacillin/Dicloxacillin n/N (%) |
Staphylococcus aureus | 73/83 (88) | 72/84 (86) |
Methicillin-resistant S. aureus | 2/3 (67) | 0/0 (-) |
Streptococcus agalactiae | 6/6 (100) | 3/6 (50) |
Streptococcus pyogenes | 18/26 (69) | 21/28 (75) |
A separate study provided additional experience with the use of ZYVOX in the treatment of methicillin-resistant Staphylococcus aureus (MRSA) infections. This was a randomized, open-label trial in hospitalized adult patients with documented or suspected MRSA infection.
One group of patients received ZYVOX I.V. Injection 600 mg every 12 hours followed by ZYVOX Tablets 600 mg every 12 hours. The other group of patients received vancomycin 1 g every 12 hours intravenously. Both groups were treated for 7 to 28 days, and could receive concomitant aztreonam or gentamicin if clinically indicated. The cure rates in microbiologically evaluable patients with MRSA skin and skin structure infection were 26/33 (79%) for linezolid-treated patients and 24/33 (73%) for vancomycin-treated patients.
Diabetic Foot Infections
Adult diabetic patients with clinically documented complicated skin and skin structure infections ("diabetic foot infections") were enrolled in a randomized (2:1 ratio), multi-center, open-label trial comparing study medications administered intravenously or orally for a total of 14 to 28 days of treatment. One group of patients received ZYVOX 600 mg every 12 hours intravenously or orally; the other group received ampicillin/sulbactam 1.5 to 3 g intravenously or amoxicillin/clavulanate 500 to 875 mg every 8 to 12 hours orally. In countries where ampicillin/sulbactam is not marketed, amoxicillin/clavulanate 500 mg to 2 g every 6 hours was used for the intravenous regimen. Patients in the comparator group could also be treated with vancomycin 1 g every 12 hours intravenously if MRSA was isolated from the foot infection. Patients in either treatment group who had Gram-negative bacilli isolated from the infection site could also receive aztreonam 1 to 2 g every 8–12 hours intravenously. All patients were eligible to receive appropriate adjunctive treatment methods, such as debridement and off-loading, as typically required in the treatment of diabetic foot infections, and most patients received these treatments. There were 241 linezolid-treated and 120 comparator-treated patients in the intent-to-treat (ITT) study population. Two hundred twelve (86%) linezolid-treated patients and 105 (85%) comparator-treated patients were clinically evaluable. In the ITT population, the cure rates were 68.5% (165/241) in linezolid-treated patients and 64% (77/120) in comparator-treated patients, where those with indeterminate and missing outcomes were considered failures. The cure rates in the clinically evaluable patients (excluding those with indeterminate and missing outcomes) were 83% (159/192) and 73% (74/101) in the linezolid- and comparator-treated patients, respectively. A critical post-hoc analysis focused on 121 linezolid-treated and 60 comparator-treated patients who had a Gram-positive pathogen isolated from the site of infection or from blood, who had less evidence of underlying osteomyelitis than the overall study population, and who did not receive prohibited antimicrobials. Based upon that analysis, the cure rates were 71% (86/121) in the linezolid-treated patients and 63% (38/60) in the comparator-treated patients. None of the above analyses were adjusted for the use of adjunctive therapies. The cure rates by pathogen for microbiologically evaluable patients are presented in Table 14.
| Cured | ||
|---|---|---|
| Pathogen | ZYVOX n/N (%) | Comparator n/N (%) |
Staphylococcus aureus | 49/63 (78) | 20/29 (69) |
Methicillin-resistant S. aureus | 12/17 (71) | 2/3 (67) |
Streptococcus agalactiae | 25/29 (86) | 9/16 (56) |
Vancomycin-Resistant Enterococcal Infections
Adult patients with documented or suspected vancomycin-resistant enterococcal infection were enrolled in a randomized, multi-center, double-blind trial comparing a high dose of ZYVOX (600 mg) with a low dose of ZYVOX (200 mg) given every 12 hours either intravenously (IV) or orally for 7 to 28 days. Patients could receive concomitant aztreonam or aminoglycosides. There were 79 patients randomized to high-dose linezolid and 66 to low-dose linezolid. The intent-to-treat (ITT) population with documented vancomycin-resistant enterococcal infection at baseline consisted of 65 patients in the high-dose arm and 52 in the low-dose arm.
The cure rates for the ITT population with documented vancomycin-resistant enterococcal infection at baseline are presented in Table 15 by source of infection. These cure rates do not include patients with missing or indeterminate outcomes. The cure rate was higher in the high-dose arm than in the low-dose arm, although the difference was not statistically significant at the 0.05 level.
| Source of Infection | Cured | |
|---|---|---|
| ZYVOX 600 mg every 12 hours n/N (%) | ZYVOX 200 mg every 12 hours n/N (%) | |
| ||
Any site | 39/58 (67) | 24/46 (52) |
Any site with associated bacteremia | 10/17 (59) | 4/14 (29) |
Bacteremia of unknown origin | 5/10 (50) | 2/7 (29) |
Skin and skin structure | 9/13 (69) | 5/5 (100) |
Urinary tract | 12/19 (63) | 12/20 (60) |
Pneumonia | 2/3 (67) | 0/1 (0) |
Other* | 11/13 (85) | 5/13 (39) |
14.2 Pediatric Patients
Infections due to Gram-positive Bacteria
A safety and efficacy study provided experience on the use of ZYVOX in pediatric patients for the treatment of nosocomial pneumonia, complicated skin and skin structure infections, and other infections due to Gram-positive bacterial pathogens, including methicillin-resistant and -susceptible Staphylococcus aureus and vancomycin-resistant Enterococcus faecium. Pediatric patients ranging in age from birth through 11 years with infections caused by the documented or suspected Gram-positive bacteria were enrolled in a randomized, open-label, comparator-controlled trial. One group of patients received ZYVOX I.V. Injection 10 mg/kg every 8 hours followed by ZYVOX for Oral Suspension 10 mg/kg every 8 hours. A second group received vancomycin 10 to 15 mg/kg intravenously every 6 to 24 hours, depending on age and renal clearance. Patients who had confirmed VRE infections were placed in a third arm of the study and received ZYVOX 10 mg/kg every 8 hours intravenously and/or orally. All patients were treated for a total of 10 to 28 days and could receive concomitant Gram-negative antibacterial drugs if clinically indicated. In the intent-to-treat (ITT) population, there were 206 patients randomized to linezolid and 102 patients randomized to vancomycin. The cure rates for ITT, MITT, and clinically evaluable patients are presented in Table 16. After the study was completed, 13 additional patients ranging from 4 days through 16 years of age were enrolled in an open-label extension of the VRE arm of the study. Table 17 provides clinical cure rates by pathogen for microbiologically evaluable patients including microbiologically evaluable patients with vancomycin-resistant Enterococcus faecium from the extension of this study.
| ITT | MITT* | Clinically Evaluable | ||||
|---|---|---|---|---|---|---|
| Population | ZYVOX n/N (%) | Vancomycin n/N (%) | ZYVOX n/N (%) | Vancomycin n/N (%) | ZYVOX n/N (%) | Vancomycin n/N (%) |
| ||||||
Any diagnosis | 150/186 (81) | 69/83 (83) | 86/108 (80) | 44/49 (90) | 106/117 (91) | 49/54 (91) |
Complicated skin and skin structure infections | 61/72 (85) | 31/34 (91) | 37/43 (86) | 22/23 (96) | 46/49 (94) | 26/27 (96) |
Nosocomial pneumonia | 13/18 (72) | 11/12 (92) | 5/6 (83) | 4/4 (100) | 7/7 (100) | 5/5 (100) |
| Microbiologically Evaluable | ||
|---|---|---|
| Pathogen | ZYVOX n/N (%) | Vancomycin n/N (%) |
| ||
Vancomycin-resistant Enterococcus faecium | 6/8 (75)* | 0/0 (-) |
Staphylococcus aureus | 36/38 (95) | 23/24 (96) |
Methicillin-resistant S. aureus | 16/17 (94) | 9/9 (100) |
Streptococcus pyogenes | 2/2 (100) | 1/2 (50) |
How Supplied/Storage and Handling
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Injection
ZYVOX I.V. Injection is available in single-dose, ready-to-use flexible plastic infusion bags in a foil laminate overwrap. The infusion bags and ports are not made with natural rubber latex. The infusion bags are available in the following package sizes:
200 mg/100 mL (2 mg/mL) linezolid × 10 | NDC 0009-5137-04 |
600 mg/300 mL (2 mg/mL) linezolid × 10 | NDC 0009-5140-04 |
16.2 Tablets
ZYVOX Tablets are available as follows:
600 mg (white, capsule-shaped, film-coated tablets debossed with "ZYV" on one side and "600" on the other)
20 tablets in HDPE bottle | NDC 0009-5138-02 |
Unit dose packages of 30 tablets | NDC 0009-5138-03 |
16.3 Oral Suspension
ZYVOX for Oral Suspension is available as a dry, white to off-white, orange-flavored granule/powder. When constituted as directed, each bottle will contain 150 mL of a suspension providing the equivalent of 100 mg of linezolid per each 5 mL. ZYVOX for Oral Suspension is supplied as follows:
100 mg/5 mL in 240 mL glass bottles | NDC 0009-5136-01 |
100 mg/5 mL in 240 mL glass bottles | NDC 0009-5136-04 |
Patient Counseling Information
17 PATIENT COUNSELING INFORMATION
Important Administration Instructions
Advise patients that ZYVOX may be taken with or without food.
Peripheral and Optic Neuropathy
Advise patients to inform their physician if they experience changes in vision while taking ZYVOX [see Warnings and Precautions (5.2)].
Serotonin Syndrome
Advise patients to inform their physician if taking serotonergic agents, including serotonin re-uptake inhibitors or other antidepressants and opioids [see Warnings and Precautions (5.3)].
Potential Interactions Producing Elevation of Blood Pressure
- •
- Advise patients to inform their physician if they have a history of hypertension.
- •
- Advise patients to avoid large quantities of foods or beverages with high tyramine content while taking ZYVOX. Foods high in tyramine content include those that may have undergone protein changes by aging, fermentation, pickling, or smoking to improve flavor, such as aged cheeses, fermented or air-dried meats, sauerkraut, soy sauce, tap beers, and red wines. The tyramine content of any protein-rich food may be increased if stored for long periods or improperly refrigerated.
- •
- Advise patients to inform their physician if taking medications containing pseudoephedrine HCl or phenylpropanolamine HCl, such as cold remedies and decongestants [see Warnings and Precautions (5.6)].
Lactic Acidosis
Advise patients to inform their physician if they experience repeated episodes of nausea or vomiting while receiving ZYVOX [see Warnings and Precautions (5.7)].
Convulsions
Advise patients to inform their physician if they have a history of seizures or convulsions [see Warnings and Precautions (5.8)].
Rhabdomyolysis
Advise patients to inform their physician if they experience signs and symptoms of rhabdomyolysis including muscle pain, tenderness or weakness and dark urine [see Warnings and Precautions (5.9)].
Hypoglycemia
Advise patients to inform their physician if they have diabetes mellitus. Hypoglycemic reactions, such as diaphoresis and tremulousness, along with low blood glucose measurements may occur when treated with linezolid. If such reactions occur, patients should contact a physician or other health professional for proper treatment [see Warnings and Precautions (5.10)].
Hyponatremia and/or SIADH
Advise patients at risk for hyponatremia to inform their physician if they experience signs and symptoms of hyponatremia and/or SIADH, including confusion, somnolence, generalized weakness, and respiratory distress [see Warnings and Precautions (5.11)].
Phenylketonuria
Advise patients with phenylketonuria (PKU) that each 5 mL of the 100 mg/5 mL ZYVOX for Oral Suspension contains 20 mg phenylalanine. The other ZYVOX formulations do not contain phenylalanine. Phenylalanine can be harmful to patients with phenylketonuria. Contact your physician or pharmacist when prescribed with ZYVOX for Oral Suspension [see Warnings and Precautions (5.12)].
Antibacterial Resistance
Patients should be counseled that antibacterial drugs including ZYVOX should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When ZYVOX is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by ZYVOX or other antibacterial drugs in the future.
Diarrhea
Diarrhea is a common problem caused by antibacterial drugs, which usually ends when the antibacterial drug is discontinued. Sometimes after starting treatment with antibacterial drugs, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibacterial drug. If this occurs, patients should contact their physician as soon as possible [see Warnings and Precautions (5.5)].
Infertility
Advise male patients that ZYVOX may reversibly impair fertility [see Use in Specific Populations (8.3)].
Other
This product's labeling may have been updated. For the most recent prescribing information, please visit www.pfizer.com.

LAB-0139-45.0
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Contact Medical Information. 9AM-5PM ET Monday to Friday; excluding holidays.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.