ALTACE® Capsules
(ramipril)
Find ALTACE® Capsules medical information:
Find ALTACE® Capsules medical information:
ALTACE® Capsules Quick Finder
17 PATIENT COUNSELING INFORMATION
17 PATIENT COUNSELING INFORMATION
Angioedema
Angioedema, including laryngeal edema, can occur with treatment with ACE inhibitors, especially following the first dose. Advise patients to immediately report any signs or symptoms suggesting angioedema (swelling of face, eyes, lips, or tongue, or difficulty in breathing) and to temporarily discontinue drug until they have consulted with the prescribing physician.
Neutropenia
Advise patients to promptly report any indication of infection (e.g., sore throat, fever), which could be a sign of neutropenia.
Symptomatic Hypotension
Inform patients that light-headedness can occur, especially during the first days of therapy, and it should be reported. Advise patients to discontinue ALTACE if syncope (fainting) occurs, and to follow up with their health care providers.
Inform patients that inadequate fluid intake or excessive perspiration, diarrhea, or vomiting while taking ALTACE can lead to an excessive fall in blood pressure, with the same consequences of lightheadedness and possible syncope.
Highlights
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ALTACE safely and effectively. See full prescribing information for ALTACE. ALTACE® (ramipril) capsules, for oral use Initial U.S. Approval: 1991 RECENT MAJOR CHANGESINDICATIONS AND USAGEALTACE is an angiotensin converting enzyme (ACE) inhibitor indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. It may be used alone or in combination with thiazide diuretics (1.1). In patients 55 years or older at high risk of developing a major cardiovascular event, ALTACE is indicated to reduce the risk of myocardial infarction, stroke, or death from cardiovascular causes (1.2). ALTACE is indicated in stable patients who have demonstrated clinical signs of congestive heart failure post-myocardial infarction (1.3). DOSAGE AND ADMINISTRATIONHypertension: Initial dose is 2.5 mg to 20 mg once daily. Adjust dosage according to blood pressure response after 2–4 weeks of treatment. The usual maintenance dose following titration is 2.5 mg to 20 mg daily as a single dose or equally divided doses (2.1). Reduction in the risk of myocardial infarction, stroke, or death from cardiovascular causes: 2.5 mg once daily for 1 week, 5 mg once daily for 3 weeks, and increased as tolerated to a maintenance dose of 10 mg once daily (2.2). Heart failure post-myocardial infarction: Starting dose of 2.5 mg twice daily. If patient becomes hypotensive at this dose, decrease dosage to 1.25 mg twice daily. Increase dose as tolerated toward a target dose of 5 mg twice daily, with dosage increases about 3 weeks apart (2.3). Dosage adjustment: See respective sections pertaining to dosage adjustment in special situations (2.5). DOSAGE FORMS AND STRENGTHSCapsule: 1.25 mg, 2.5 mg, 5 mg, 10 mg (3) CONTRAINDICATIONSAngioedema related to previous treatment with an ACE inhibitor, or a history of hereditary or idiopathic angioedema (4). ALTACE is contraindicated in combination with a neprilysin inhibitor (e.g., sacubitril). Do not administer ALTACE within 36 hours of switching to or from sacubitril/valsartan, a neprilysin inhibitor (4). Do not co-administer aliskiren with ALTACE in patients with diabetes (4). WARNINGS AND PRECAUTIONSAngioedema, increased risk in patients with a prior history (5.1) Hypotension and hyperkalemia (5.5, 5.8) Renal impairment: monitor renal function during therapy (5.3) Avoid concomitant use of an ACE inhibitor and angiotensin blocker (5.7) Rare cholestatic jaundice and hepatic failure (5.2) Rare neutropenia and agranulocytosis (5.4) ADVERSE REACTIONSThe most common adverse reactions in patients with hypertension included headache, dizziness, fatigue, and cough (6.1). To report SUSPECTED ADVERSE REACTIONS, contact Pfizer at 1-800-438-1985 or the FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSDiuretics: Possibility of excessive hypotension (7.1). Lithium: Use with caution (7.4). Gold: Nitritoid reactions have been reported (7.5). NSAIDS use may lead to increased risk of renal impairment and loss of antihypertensive effect (7.6). mTOR inhibitor or neprilysin inhibitor use may increase angioedema risk (7.7). USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 11/2023 |
Boxed Warning
Indications and Usage
1 INDICATIONS AND USAGE
1.1 Hypertension
ALTACE is indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes including this drug.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program's Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
ALTACE may be used alone or in combination with thiazide diuretics.
1.2 Reduction in the Risk of Myocardial Infarction, Stroke, and Death from Cardiovascular Causes
ALTACE is indicated in patients 55 years or older at high risk of developing a major cardiovascular event because of a history of coronary artery disease, stroke, peripheral vascular disease, or diabetes that is accompanied by at least one other cardiovascular risk factor (hypertension, elevated total cholesterol levels, low HDL levels, cigarette smoking, or documented microalbuminuria), to reduce the risk of myocardial infarction, stroke, or death from cardiovascular causes. ALTACE can be used in addition to other needed treatment (such as antihypertensive, antiplatelet, or lipid-lowering therapy) [see Clinical Studies (14.2)].
1.3 Heart Failure Post-Myocardial Infarction
ALTACE is indicated in stable patients who have demonstrated clinical signs of congestive heart failure within the first few days after sustaining acute myocardial infarction. Administration of ALTACE to such patients has been shown to decrease the risk of death (principally cardiovascular death) and to decrease the risks of failure-related hospitalization and progression to severe/resistant heart failure [see Clinical Studies (14.3)].
Dosage and Administration
2 DOSAGE AND ADMINISTRATION
2.1 Hypertension
The recommended initial dose for patients not receiving a diuretic is 2.5 mg once a day. Adjust dose according to blood pressure response. The usual maintenance dosage range is 2.5 mg to 20 mg per day administered as a single dose or in two equally divided doses. In some patients treated once daily, the antihypertensive effect may diminish toward the end of the dosing interval. In such patients, consider an increase in dosage or twice daily administration. If blood pressure is not controlled with ALTACE alone, a diuretic can be added.
2.2 Reduction in Risk of Myocardial Infarction, Stroke, and Death from Cardiovascular Causes
Initiate dosing at 2.5 mg once daily for 1 week, 5 mg once daily for the next 3 weeks, and then increase as tolerated, to a maintenance dose of 10 mg once daily. If the patient is hypertensive or recently post-myocardial infarction, ALTACE can also be given as a divided dose.
2.3 Heart Failure Post-Myocardial Infarction
For the treatment of post-myocardial infarction patients who have shown signs of congestive heart failure, the recommended starting dose of ALTACE is 2.5 mg twice daily (5 mg per day). A patient who becomes hypotensive at this dose may be switched to 1.25 mg twice daily. After one week at the starting dose, increase dose (if tolerated) toward a target dose of 5 mg twice daily, with dosage increases being about 3 weeks apart.
After the initial dose of ALTACE, observe the patient under medical supervision for at least two hours and until blood pressure has stabilized for at least an additional hour. If possible, reduce the dose of any concomitant diuretic as this may diminish the likelihood of hypotension. The appearance of hypotension after the initial dose of ALTACE does not preclude subsequent careful dose titration with the drug, following effective management of the hypotension [see Warnings and Precautions (5.5), Drug Interactions (7.1)].
2.4 General Dosing Information
Generally, swallow ALTACE capsules whole. The ALTACE capsule can also be opened and the contents sprinkled on a small amount (about 4 oz.) of applesauce or mixed in 4 oz. (120 mL) of water or apple juice. To be sure that ramipril is not lost when such a mixture is used, consume the mixture in its entirety. The described mixtures can be pre-prepared and stored for up to 24 hours at room temperature or up to 48 hours under refrigeration.
Concomitant administration of ALTACE with potassium supplements, potassium salt substitutes, or potassium-sparing diuretics can lead to increases of serum potassium [see Warnings and Precautions (5.8)].
2.5 Dosage Adjustment
Renal Impairment
Establish baseline renal function in patients initiating ALTACE. Usual regimens of therapy with ALTACE may be followed in patients with estimated creatinine clearance >40 mL/min. However, in patients with worse impairment, 25% of the usual dose of ramipril is expected to produce full therapeutic levels of ramiprilat [see Use in Specific Populations (8.6)].
Hypertension
For patients with hypertension and renal impairment, the recommended initial dose is 1.25 mg ALTACE once daily. Dosage may be titrated upward until blood pressure is controlled or to a maximum total daily dose of 5 mg.
Heart Failure Post-Myocardial Infarction
For patients with heart failure and renal impairment, the recommended initial dose is 1.25 mg ALTACE once daily. The dose may be increased to 1.25 mg twice daily, and up to a maximum dose of 2.5 mg twice daily depending on clinical response and tolerability.
Volume Depletion or Renal Artery Stenosis
Blood pressure decreases associated with any dose of ALTACE depend, in part, on the presence or absence of volume depletion (e.g., past and current diuretic use) or the presence or absence of renal artery stenosis. If such circumstances are suspected to be present, initiate dosing at 1.25 mg once daily. Adjust dosage according to blood pressure response.
Dosage Forms and Strengths
Contraindications
4 CONTRAINDICATIONS
ALTACE is contraindicated in patients who are hypersensitive to this product or any other ACE inhibitor (e.g., a patient who has experienced angioedema during therapy with any other ACE inhibitor).
ALTACE is contraindicated in combination with a neprilysin inhibitor (e.g., sacubitril). Do not administer ALTACE within 36 hours of switching to or from sacubitril/valsartan, a neprilysin inhibitor [see Warnings and Precautions (5.1)].
Do not co-administer ALTACE with aliskiren:
- •
- in patients with diabetes
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylactoid and Possibly Related Reactions
Presumably because drugs that act directly on the renin-angiotensin-aldosterone system (e.g., ACE inhibitors) affect the metabolism of eicosanoids and polypeptides, including endogenous bradykinin, patients receiving these drugs (including ALTACE) may be subject to a variety of adverse reactions, some of them serious.
Angioedema
Head and Neck Angioedema
Patients with a history of angioedema unrelated to ACE inhibitor therapy may be at increased risk of angioedema while receiving an ACE inhibitor.
Angioedema of the face, extremities, lips, tongue, glottis, and larynx has been reported in patients treated with ACE inhibitors. Angioedema associated with laryngeal edema can be fatal. If laryngeal stridor or angioedema of the face, tongue, or glottis occurs, discontinue treatment with ALTACE and institute appropriate therapy immediately. Where there is involvement of the tongue, glottis, or larynx likely to cause airway obstruction, administer appropriate therapy (e.g., subcutaneous epinephrine solution 1:1000 [0.3 mL to 0.5 mL]) promptly [see Adverse Reactions (6)].
In considering the use of ALTACE, note that in controlled clinical trials ACE inhibitors cause a higher rate of angioedema in Black patients than in non-Black patients.
In a large U.S. post-marketing study, angioedema (defined as reports of angio, face, larynx, tongue, or throat edema) was reported in 3/1523 (0.20%) Black patients and in 8/8680 (0.09%) non-Black patients. These rates were not different statistically.
Patients taking concomitant mammalian target of rapamycin (mTOR) inhibitor (e.g. temsirolimus) therapy or a neprilysin inhibitor may be at increased risk for angioedema [see Drug Interactions (7.7)].
Intestinal Angioedema
Intestinal angioedema has been reported in patients treated with ACE inhibitors. These patients presented with abdominal pain (with or without nausea or vomiting); in some cases there was no prior history of facial angioedema and C-1 esterase levels were normal. The angioedema was diagnosed by procedures including abdominal CT scan or ultrasound, or at surgery, and symptoms resolved after stopping the ACE inhibitor. Include intestinal angioedema in the differential diagnosis of patients on ACE inhibitors presenting with abdominal pain.
Anaphylactoid Reactions During Desensitization
Two patients undergoing desensitizing treatment with hymenoptera venom while receiving ACE inhibitors sustained life-threatening anaphylactoid reactions. In the same patients, these reactions were avoided when ACE inhibitors were temporarily withheld, but they reappeared upon inadvertent rechallenge.
Anaphylactoid Reactions During Membrane Exposure
Anaphylactoid reactions have been reported in patients dialyzed with high-flux membranes and treated concomitantly with an ACE inhibitor. Anaphylactoid reactions have also been reported in patients undergoing low-density lipoprotein apheresis with dextran sulfate absorption.
5.2 Hepatic Failure and Impaired Liver Function
Rarely, ACE inhibitors, including ALTACE, have been associated with a syndrome that starts with cholestatic jaundice and progresses to fulminant hepatic necrosis and sometimes death. The mechanism of this syndrome is not understood. Discontinue ALTACE if patient develops jaundice or marked elevations of hepatic enzymes.
As ramipril is primarily metabolized by hepatic esterases to its active moiety, ramiprilat, patients with impaired liver function could develop markedly elevated plasma levels of ramipril. No formal pharmacokinetic studies have been carried out in hypertensive patients with impaired liver function.
5.3 Renal Impairment
As a consequence of inhibiting the renin-angiotensin-aldosterone system, changes in renal function may be anticipated in susceptible individuals. In patients with severe congestive heart failure whose renal function may depend on the activity of the renin-angiotensin-aldosterone system, treatment with ACE inhibitors, including ALTACE, may be associated with oliguria or progressive azotemia and rarely with acute renal failure or death.
In hypertensive patients with unilateral or bilateral renal artery stenosis, increases in blood urea nitrogen and serum creatinine may occur. Experience with another ACE inhibitor suggests that these increases would be reversible upon discontinuation of ALTACE and/or diuretic therapy. In such patients, monitor renal function during the first few weeks of therapy. Some hypertensive patients with no apparent pre-existing renal vascular disease have developed increases in blood urea nitrogen and serum creatinine, usually minor and transient, especially when ALTACE has been given concomitantly with a diuretic. This is more likely to occur in patients with pre-existing renal impairment. Dosage reduction of ALTACE and/or discontinuation of the diuretic may be required.
5.4 Neutropenia and Agranulocytosis
In rare instances, treatment with ACE inhibitors may be associated with mild reductions in red blood cell count and hemoglobin content, blood cell or platelet counts. In isolated cases, agranulocytosis, pancytopenia, and bone marrow depression may occur. Hematological reactions to ACE inhibitors are more likely to occur in patients with collagen-vascular disease (e.g., systemic lupus erythematosus, scleroderma) and renal impairment. Consider monitoring white blood cell counts in patients with collagen-vascular disease, especially if the disease is associated with impaired renal function.
5.5 Hypotension
General Considerations
ALTACE can cause symptomatic hypotension, after either the initial dose or a later dose when the dosage has been increased. Like other ACE inhibitors, ALTACE, has been only rarely associated with hypotension in uncomplicated hypertensive patients. Symptomatic hypotension is most likely to occur in patients who have been volume- and/or salt-depleted as a result of prolonged diuretic therapy, dietary salt restriction, dialysis, diarrhea, or vomiting. Correct volume- and salt-depletion before initiating therapy with ALTACE.
If excessive hypotension occurs, place the patient in a supine position and, if necessary, treat with intravenous infusion of physiological saline. ALTACE treatment usually can be continued following restoration of blood pressure and volume.
Heart Failure Post-Myocardial Infarction
In patients with heart failure post-myocardial infarction who are currently being treated with a diuretic, symptomatic hypotension occasionally can occur following the initial dose of ALTACE. If the initial dose of 2.5 mg ALTACE cannot be tolerated, use an initial dose of 1.25 mg ALTACE to avoid excessive hypotension. Consider reducing the dose of concomitant diuretic to decrease the incidence of hypotension.
Congestive Heart Failure
In patients with congestive heart failure, with or without associated renal insufficiency, ACE inhibitor therapy may cause excessive hypotension, which may be associated with oliguria or azotemia and rarely, with acute renal failure and death. In such patients, initiate ALTACE therapy under close medical supervision and follow patients closely for the first 2 weeks of treatment and whenever the dose of ALTACE or diuretic is increased.
Surgery and Anesthesia
In patients undergoing surgery or during anesthesia with agents that produce hypotension, ramipril may block angiotensin II formation that would otherwise occur secondary to compensatory renin release. Hypotension that occurs as a result of this mechanism can be corrected by volume expansion.
5.6 Fetal Toxicity
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue ALTACE as soon as possible [see Use in Specific Populations (8.1)].
5.7 Dual Blockade of the Renin-Angiotensin System
Dual blockade of the RAS with angiotensin receptor blockers, ACE inhibitors, or aliskiren is associated with increased risks of hypotension, hyperkalemia, and changes in renal function (including acute renal failure) compared to monotherapy. Most patients receiving the combination of two RAS inhibitors do not obtain any additional benefit compared to monotherapy. In general, avoid combined use of RAS inhibitors. Closely monitor blood pressure, renal function and electrolytes in patients on ALTACE and other agents that affect the RAS.
Telmisartan
The ONTARGET trial enrolled 25,620 patients >55 years old with atherosclerotic disease or diabetes with end-organ damage, randomized them to telmisartan only, ramipril only, or the combination, and followed them for a median of 56 months. Patients receiving the combination of telmisartan and ramipril did not obtain any benefit in the composite endpoint of cardiovascular death, MI, stroke and heart failure hospitalization compared to monotherapy, but experienced an increased incidence of clinically important renal dysfunction (death, doubling of serum creatinine, or dialysis) compared with groups receiving telmisartan alone or ramipril alone. Concomitant use of telmisartan and ramipril is not recommended.
5.8 Hyperkalemia
In clinical trials with ALTACE, hyperkalemia (serum potassium >5.7 mEq/L) occurred in approximately 1% of hypertensive patients receiving ALTACE. In most cases, these were isolated values, which resolved despite continued therapy. None of these patients were discontinued from the trials because of hyperkalemia. Risk factors for the development of hyperkalemia include renal insufficiency, diabetes mellitus, and the concomitant use of other drugs that raise serum potassium levels. Monitor serum potassium in such patients [see Drug Interactions (7.2)].
5.9 Cough
Presumably caused by inhibition of the degradation of endogenous bradykinin, persistent nonproductive cough has been reported with all ACE inhibitors, always resolving after discontinuation of therapy. Consider the possibility of angiotensin converting enzyme inhibitor induced-cough in the differential diagnosis of cough.
Adverse Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Hypertension
ALTACE has been evaluated for safety in over 4000 patients with hypertension; of these, 1230 patients were studied in U.S. controlled trials, and 1107 were studied in foreign controlled trials. Almost 700 of these patients were treated for at least one year. The overall incidence of reported adverse events was similar in ALTACE and placebo patients. The most frequent clinical side effects (possibly or probably related to study drug) reported by patients receiving ALTACE in placebo-controlled trials were: headache (5.4%), dizziness (2.2%), and fatigue or asthenia (2.0%), but only the last one was more common in ALTACE patients than in patients given placebo. Generally the side effects were mild and transient, and there was no relation to total dosage within the range of 1.25 mg–20 mg. Discontinuation of therapy because of a side effect was required in approximately 3% of U.S. patients treated with ALTACE. The most common reasons for discontinuation were: cough (1.0%), dizziness (0.5%), and impotence (0.4%).
Of observed side effects considered possibly or probably related to study drug that occurred in U.S. placebo-controlled trials in more than 1% of patients treated with ALTACE, only asthenia (fatigue) was more common on ALTACE than placebo (2% [n=13/651] vs. 1% [n=2/286], respectively).
In placebo-controlled trials, there was also an excess of upper respiratory infection and flu syndrome in the ALTACE group, not attributed at that time to ramipril. As these studies were carried out before the relationship of cough to ACE inhibitors was recognized, some of these events may represent ramipril-induced cough. In a later 1-year study, increased cough was seen in almost 12% of ALTACE patients, with about 4% of patients requiring discontinuation of treatment.
Reduction in the Risk of Myocardial Infarction, Stroke, and Death from Cardiovascular Causes
HOPE Study
Safety data in the Heart Outcomes Prevention Evaluation (HOPE) study were collected as reasons for discontinuation or temporary interruption of treatment. The incidence of cough was similar to that seen in the Acute Infarction Ramipril Efficacy (AIRE) trial. The rate of angioedema was the same as in previous clinical trials [see Warnings and Precautions (5.1)].
| Placebo (N=4652) | ALTACE (N=4645) | |
|---|---|---|
Discontinuation at any time | 32% | 34% |
Permanent discontinuation | 28% | 29% |
Reasons for stopping | ||
Cough | 2% | 7% |
Hypotension or dizziness | 1.5% | 1.9% |
Angioedema | 0.1% | 0.3% |
Heart Failure Post-Myocardial Infarction
AIRE Study
Adverse reactions (except laboratory abnormalities) considered possibly/probably related to study drug that occurred in more than 1% of patients and more frequently on ALTACE are shown below. The incidences are from the AIRE study. The follow-up time was between 6 and 46 months for this study.
| Adverse Event | Placebo (N=982) | ALTACE (N=1004) |
|---|---|---|
Hypotension | 5% | 11% |
Cough increased | 4% | 8% |
Dizziness | 3% | 4% |
Angina pectoris | 2% | 3% |
Nausea | 1% | 2% |
Postural hypotension | 1% | 2% |
Syncope | 1% | 2% |
Vomiting | 0.5% | 2% |
Vertigo | 0.7% | 2% |
Abnormal kidney function | 0.5% | 1% |
Diarrhea | 0.4% | 1% |
Other Adverse Reactions
Other adverse reactions reported in controlled clinical trials (in less than 1% of ALTACE patients), or rarer events seen in post-marketing experience, include the following (in some, a causal relationship to drug is uncertain):
Body as a whole: Anaphylactoid reactions [see Warnings and Precautions (5.1)].
Cardiovascular: Symptomatic hypotension (reported in 0.5% of patients in U.S. trials) [see Warnings and Precautions (5.5)], syncope, and palpitations.
Hematologic: Pancytopenia, hemolytic anemia, and thrombocytopenia.
Decreases in hemoglobin or hematocrit (a low value and a decrease of 5 g/dL or 5%, respectively) were rare, occurring in 0.4% of patients receiving ALTACE alone and in 1.5% of patients receiving ALTACE plus a diuretic.
Renal: Acute renal failure. Some hypertensive patients with no apparent pre-existing renal disease have developed minor, usually transient, increases in blood urea nitrogen and serum creatinine when taking ALTACE, particularly when ALTACE was given concomitantly with a diuretic [see Warnings and Precautions (5.3)].
Angioneurotic edema: Angioneurotic edema has been reported in 0.3% of patients in U.S. clinical trials of ALTACE [see Warnings and Precautions (5.1) ].
Gastrointestinal: Hepatic failure, hepatitis, jaundice, pancreatitis, abdominal pain (sometimes with enzyme changes suggesting pancreatitis), anorexia, constipation, diarrhea, dry mouth, dyspepsia, dysphagia, gastroenteritis, increased salivation, and taste disturbance.
Dermatologic: Apparent hypersensitivity reactions (manifested by urticaria, pruritus, or rash, with or without fever), photosensitivity, purpura, onycholysis, pemphigus, pemphigoid, erythema multiforme, toxic epidermal necrolysis, and Stevens-Johnson syndrome.
Neurologic and Psychiatric: Anxiety, amnesia, convulsions, depression, hearing loss, insomnia, nervousness, neuralgia, neuropathy, paresthesia, somnolence, tinnitus, tremor, vertigo, and vision disturbances.
Miscellaneous: As with other ACE inhibitors, a symptom complex has been reported which may include a positive ANA, an elevated erythrocyte sedimentation rate, arthralgia/arthritis, myalgia, fever, vasculitis, eosinophilia, photosensitivity, rash and other dermatologic manifestations. Additionally, as with other ACE inhibitors, eosinophilic pneumonitis has been reported.
Other: Arthralgia, arthritis, dyspnea, edema, epistaxis, impotence, increased sweating, malaise, myalgia, and weight gain.
6.2 Post-Marketing Experience
In addition to adverse reactions reported from clinical trials, there have been rare reports of hypoglycemia reported during ALTACE therapy when given to patients concomitantly taking oral hypoglycemic agents or insulin. The causal relationship is unknown.
6.3 Clinical Laboratory Test Findings
Creatinine and Blood Urea Nitrogen: Increases in creatinine levels occurred in 1.2% of patients receiving ALTACE alone, and in 1.5% of patients receiving ALTACE and a diuretic. Increases in blood urea nitrogen levels occurred in 0.5% of patients receiving ALTACE alone and in 3% of patients receiving ALTACE with a diuretic. None of these increases required discontinuation of treatment. Increases in these laboratory values are more likely to occur in patients with renal insufficiency or those pretreated with a diuretic and, based on experience with other ACE inhibitors, would be expected to be especially likely in patients with renal artery stenosis [see Warnings and Precautions (5.3)]. As ramipril decreases aldosterone secretion, elevation of serum potassium can occur. Use potassium supplements and potassium sparing diuretics with caution, and monitor the patient's serum potassium frequently [see Warnings and Precautions (5.8)].
Hemoglobin and Hematocrit: Decreases in hemoglobin or hematocrit (a low value and a decrease of 5 g/dL or 5%, respectively) were rare, occurring in 0.4% of patients receiving ALTACE alone and in 1.5% of patients receiving ALTACE plus a diuretic. No US patients discontinued treatment because of decreases in hemoglobin or hematocrit.
Other (causal relationships unknown): Clinically important changes in standard laboratory tests were rarely associated with ALTACE administration. Elevations of liver enzymes, serum bilirubin, uric acid, and blood glucose have been reported, as have cases of hyponatremia and scattered incidents of leucopenia, eosinophilia, and proteinuria. In US trials, less than 0.2% of patients discontinued treatment for laboratory abnormalities; all of these were cases of proteinuria or abnormal liver-function tests.
Drug Interactions
7 DRUG INTERACTIONS
7.1 Diuretics
Patients on diuretics, especially those in whom diuretic therapy was recently instituted, may occasionally experience an excessive reduction of blood pressure after initiation of therapy with ALTACE. The possibility of hypotensive effects with ALTACE can be minimized by either decreasing or discontinuing the diuretic or increasing the salt intake prior to initiation of treatment with ALTACE. If this is not possible, reduce the starting dose [see Dosage and Administration (2)].
7.2 Agents Increasing Serum Potassium
Coadministration of ALTACE with other drugs that raise serum potassium levels may result in hyperkalemia. Monitor serum potassium in such patients.
7.3 Other Agents Affecting RAS
In general, avoid combined use of RAS inhibitors. [see Warnings and Precautions (5.7)]. Do not co-administer aliskiren with ALTACE in patients with diabetes [see Contraindications (4)].
7.4 Lithium
Increased serum lithium levels and symptoms of lithium toxicity have been reported in patients receiving ACE inhibitors during therapy with lithium; therefore, frequent monitoring of serum lithium levels is recommended. If a diuretic is also used, the risk of lithium toxicity may be increased.
7.5 Gold
Nitritoid reactions (symptoms include facial flushing, nausea, vomiting and hypotension) have been reported rarely in patients on therapy with injectable gold (sodium aurothiomalate) and concomitant ACE inhibitor therapy including ALTACE.
7.6 Non-Steroidal Anti-Inflammatory Agents including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, co-administration of NSAIDs, including selective COX-2 inhibitors, with ACE inhibitors, including ramipril, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving ramipril and NSAID therapy.
The antihypertensive effect of ACE inhibitors, including ramipril, may be attenuated by NSAIDs.
7.7 mTOR Inhibitors or Other Drugs Known to Cause Angioedema
Patients taking concomitant mTOR inhibitor (e.g. temsirolimus) therapy or a neprilysin inhibitor may be at increased risk for angioedema [see Warnings and Precautions (5.1)].
Use in Specific Populations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue ALTACE as soon as possible. These adverse outcomes are usually associated with use of these drugs in the second and third trimester of pregnancy. Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. Appropriate management of maternal hypertension during pregnancy is important to optimize outcomes for both mother and fetus.
In the unusual case that there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system for a particular patient, apprise the mother of the potential risk to the fetus. Perform serial ultrasound examinations to assess the intra-amniotic environment. If oligohydramnios is observed, discontinue ALTACE unless it is considered life-saving for the mother. Fetal testing may be appropriate, based on the week of pregnancy. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. Closely observe infants with histories of in utero exposure to ALTACE for hypotension, oliguria, and hyperkalemia [see Use in Specific Populations (8.4)].
8.3 Nursing Mothers
Ingestion of a single 10 mg oral dose of ALTACE resulted in undetectable amounts of ramipril and its metabolites in breast milk. However, because multiple doses may produce low milk concentrations that are not predictable from a single dose, do not use ALTACE in nursing mothers.
8.4 Pediatric Use
Neonates with a history of in utero exposure to ALTACE:
If oliguria or hypotension occurs, direct attention toward support of blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and/or substituting for disordered renal function. Ramipril, which crosses the placenta, can be removed from the neonatal circulation by these means, but limited experience has not shown that such removal is central to the treatment of these infants.
Safety and effectiveness in pediatric patients have not been established. Irreversible kidney damage has been observed in very young rats given a single dose of ALTACE.
8.5 Geriatric Use
Of the total number of patients who received ALTACE in U.S. clinical studies of ALTACE, 11.0% were ≥65 years of age while 0.2% were ≥75 years of age. No overall differences in effectiveness or safety were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but a greater sensitivity of some older individuals cannot be ruled out.
One pharmacokinetic study conducted in hospitalized elderly patients indicated that peak ramiprilat levels and area under the plasma concentration-time curve (AUC) for ramiprilat are higher in older patients.
8.6 Renal Impairment
A single-dose pharmacokinetic study was conducted in hypertensive patients with varying degrees of renal impairment who received a single 10 mg dose of ramipril. Patients were stratified into four groups based on initial estimates of creatinine clearance: normal (>80 mL/min), mild impairment (40–80 mL/min), moderate impairment (15–40 mL/min), and severe impairment (<15 mL/min). On average, the AUC0–24h for ramiprilat was approximately 1.7-fold higher, 3.0-fold higher, and 3.2-fold higher in the groups with mild, moderate, and severe renal impairment, respectively, compared to the group with normal renal function. Overall, the results suggest that the starting dose of ramipril should be adjusted downward in patients with moderate-to-severe renal impairment.
Overdosage
10 OVERDOSAGE
Single oral doses of ramipril in rats and mice of 10 g/kg–11 g/kg resulted in significant lethality. In dogs, oral doses as high as 1 g/kg induced only mild gastrointestinal distress. Limited data on human overdosage are available. The most likely clinical manifestations would be symptoms attributable to hypotension.
Laboratory determinations of serum levels of ramipril and its metabolites are not widely available, and such determinations have, in any event, no established role in the management of ramipril overdose.
No data are available to suggest physiological maneuvers (e.g., maneuvers to change the pH of the urine) that might accelerate elimination of ramipril and its metabolites. Similarly, it is not known which, if any, of these substances can be effectively removed from the body by hemodialysis.
Angiotensin II could presumably serve as a specific antagonist-antidote in the setting of ramipril overdose, but angiotensin II is essentially unavailable outside of scattered research facilities. Because the hypotensive effect of ramipril is achieved through vasodilation and effective hypovolemia, it is reasonable to treat ramipril overdose by infusion of normal saline solution.
Description
11 DESCRIPTION
Ramipril is a 2-aza-bicyclo [3.3.0]-octane-3-carboxylic acid derivative. It is a white, crystalline substance soluble in polar organic solvents and buffered aqueous solutions. Ramipril melts between 105°–112°C.
The CAS Registry Number is 87333-19-5. Ramipril's chemical name is (2S,3aS,6aS)-1[(S)-N-[(S)-1-Carboxy-3-phenylpropyl] alanyl] octahydrocyclopenta [b]pyrrole-2-carboxylic acid, 1-ethyl ester.
The inactive ingredients present are pregelatinized starch NF, gelatin, and titanium dioxide. The 1.25 mg capsule shell contains yellow iron oxide, the 2.5 mg capsule shell contains D&C yellow #10 and FD&C red #40, the 5 mg capsule shell contains FD&C blue #1 and FD&C red #40, and the 10 mg capsule shell contains FD&C blue #1.
The structural formula for ramipril is:
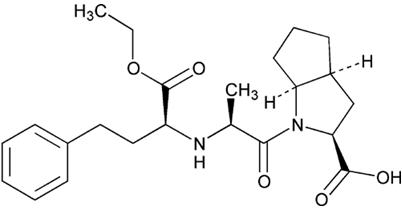
Its empirical formula is C23H32N2O5 and its molecular weight is 416.5.
Ramiprilat, the diacid metabolite of ramipril, is a non-sulfhydryl ACE inhibitor. Ramipril is converted to ramiprilat by hepatic cleavage of the ester group.
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ramipril and ramiprilat inhibit ACE in human subjects and animals. Angiotensin converting enzyme is a peptidyl dipeptidase that catalyzes the conversion of angiotensin I to the vasoconstrictor substance, angiotensin II. Angiotensin II also stimulates aldosterone secretion by the adrenal cortex. Inhibition of ACE results in decreased plasma angiotensin II, which leads to decreased vasopressor activity and to decreased aldosterone secretion. The latter decrease may result in a small increase of serum potassium. In hypertensive patients with normal renal function treated with ALTACE alone for up to 56 weeks, approximately 4% of patients during the trial had an abnormally high serum potassium and an increase from baseline greater than 0.75 mEq/L, and none of the patients had an abnormally low potassium and a decrease from baseline greater than 0.75 mEq/L. In the same study, approximately 2% of patients treated with ALTACE and hydrochlorothiazide for up to 56 weeks had abnormally high potassium values and an increase from baseline of 0.75 mEq/L or greater; and approximately 2% had abnormally low values and decreases from baseline of 0.75 mEq/L or greater [see Warnings and Precautions (5.8)]. Removal of angiotensin II negative feedback on renin secretion leads to increased plasma renin activity.
The effect of ramipril on hypertension appears to result at least in part from inhibition of both tissue and circulating ACE activity, thereby reducing angiotensin II formation in tissue and plasma.
Angiotensin converting enzyme is identical to kininase, an enzyme that degrades bradykinin. Whether increased levels of bradykinin, a potent vasopressor peptide, play a role in the therapeutic effects of ALTACE remains to be elucidated.
While the mechanism through which ALTACE lowers blood pressure is believed to be primarily suppression of the renin-angiotensin-aldosterone system, ALTACE has an antihypertensive effect even in patients with low-renin hypertension. Although ALTACE was antihypertensive in all races studied, Black hypertensive patients (usually a low-renin hypertensive population) had a blood pressure lowering response to monotherapy, albeit a smaller average response, than non-Black patients.
12.2 Pharmacodynamics
Single doses of ramipril of 2.5 mg–20 mg produce approximately 60%–80% inhibition of ACE activity 4 hours after dosing with approximately 40%–60% inhibition after 24 hours. Multiple oral doses of ramipril of 2.0 mg or more cause plasma ACE activity to fall by more than 90% 4 hours after dosing, with over 80% inhibition of ACE activity remaining 24 hours after dosing. The more prolonged effect of even small multiple doses presumably reflects saturation of ACE binding sites by ramiprilat and relatively slow release from those sites.
12.3 Pharmacokinetics
Absorption
Following oral administration of ALTACE, peak plasma concentrations (Cmax) of ramipril are reached within 1 hour. The extent of absorption is at least 50%–60%, and is not significantly influenced by the presence of food in the gastrointestinal tract, although the rate of absorption is reduced.
In a trial in which subjects received ALTACE capsules or the contents of identical capsules dissolved in water, dissolved in apple juice, or suspended in applesauce, serum ramiprilat levels were essentially unrelated to the use or non-use of the concomitant liquid or food.
Distribution
Cleavage of the ester group (primarily in the liver) converts ramipril to its active diacid metabolite, ramiprilat. Peak plasma concentrations of ramiprilat are reached 2–4 hours after drug intake. The serum protein binding of ramipril is about 73% and that of ramiprilat about 56%; in vitro, these percentages are independent of concentration over the range of 0.01 µg/mL–10 µg/mL.
Metabolism
Ramipril is almost completely metabolized to ramiprilat, which has about 6 times the ACE inhibitory activity of ramipril, and to the diketopiperazine ester, the diketopiperazine acid, and the glucuronides of ramipril and ramiprilat, all of which are inactive.
Plasma concentrations of ramipril and ramiprilat increase with increased dose, but are not strictly dose-proportional. The 24-hour AUC for ramiprilat, however, is dose-proportional over the 2.5 mg–20 mg dose range. The absolute bioavailabilities of ramipril and ramiprilat were 28% and 44%, respectively, when 5 mg of oral ramipril was compared with the same dose of ramipril given intravenously.
After once-daily dosing, steady-state plasma concentrations of ramiprilat are reached by the fourth dose. Steady-state concentrations of ramiprilat are somewhat higher than those seen after the first dose of ALTACE, especially at low doses (2.5 mg), but the difference is clinically insignificant.
Plasma concentrations of ramiprilat decline in a triphasic manner (initial rapid decline, apparent elimination phase, terminal elimination phase). The initial rapid decline, which represents distribution of the drug into a large peripheral compartment and subsequent binding to both plasma and tissue ACE, has a half-life of 2–4 hours. Because of its potent binding to ACE and slow dissociation from the enzyme, ramiprilat shows two elimination phases. The apparent elimination phase corresponds to the clearance of free ramiprilat and has a half-life of 9–18 hours. The terminal elimination phase has a prolonged half-life (>50 hours) and probably represents the binding/dissociation kinetics of the ramiprilat/ACE complex. It does not contribute to the accumulation of the drug. After multiple daily doses of ALTACE 5 mg–10 mg, the half-life of ramiprilat concentrations within the therapeutic range was 13–17 hours.
In patients with creatinine clearance <40 mL/min/1.73 m2, peak levels of ramiprilat are approximately doubled, and trough levels may be as much as quintupled. In multiple-dose regimens, the total exposure to ramiprilat (AUC) in these patients is 3–4 times as large as it is in patients with normal renal function who receive similar doses.
In patients with impaired liver function, the metabolism of ramipril to ramiprilat appears to be slowed, possibly because of diminished activity of hepatic esterases, and plasma ramipril levels in these patients are increased about 3-fold. Peak concentrations of ramiprilat in these patients, however, are not different from those seen in subjects with normal hepatic function, and the effect of a given dose on plasma ACE activity does not vary with hepatic function.
Excretion
After oral administration of ramipril, about 60% of the parent drug and its metabolites are eliminated in the urine, and about 40% is found in the feces. Drug recovered in the feces may represent both biliary excretion of metabolites and/or unabsorbed drug, however the proportion of a dose eliminated by the bile has not been determined. Less than 2% of the administered dose is recovered in urine as unchanged ramipril.
The urinary excretion of ramipril, ramiprilat, and their metabolites is reduced in patients with impaired renal function. Compared to normal subjects, patients with creatinine clearance <40 mL/min/1.73 m2 had higher peak and trough ramiprilat levels and slightly longer times to peak concentrations.
Nonclinical Toxicology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No evidence of a tumorigenic effect was found when ramipril was given by gavage to rats for up to 24 months at doses of up to 500 mg/kg/day or to mice for up to 18 months at doses of up to 1000 mg/kg/day. (For either species, these doses are about 200 times the maximum recommended human dose when compared on the basis of body surface area.) No mutagenic activity was detected in the Ames test in bacteria, the micronucleus test in mice, unscheduled DNA synthesis in a human cell line, or a forward gene-mutation assay in a Chinese hamster ovary cell line. Several metabolites and degradation products of ramipril were also negative in the Ames test. A study in rats with dosages as great as 500 mg/kg/day did not produce adverse effects on fertility.
No teratogenic effects of ramipril were seen in studies of pregnant rats, rabbits, and cynomolgus monkeys. On a body surface area basis, the doses used were up to approximately 400 times (in rats and monkeys) and 2 times (in rabbits) the recommended human dose.
Clinical Studies
14 CLINICAL STUDIES
14.1 Hypertension
ALTACE has been compared with other ACE inhibitors, beta-blockers, and thiazide diuretics as monotherapy for hypertension. It was approximately as effective as other ACE inhibitors and as atenolol.
Administration of ALTACE to patients with mild to moderate hypertension results in a reduction of both supine and standing blood pressure to about the same extent with no compensatory tachycardia. Symptomatic postural hypotension is infrequent, although it can occur in patients who are salt- and/or volume-depleted [see Warnings and Precautions (5.5)]. Use of ALTACE in combination with thiazide diuretics gives a blood pressure lowering effect greater than that seen with either agent alone.
In single-dose studies, doses of 5 mg–20 mg of ALTACE lowered blood pressure within 1–2 hours, with peak reductions achieved 3–6 hours after dosing. The antihypertensive effect of a single dose persisted for 24 hours. In longer term (4–12 weeks) controlled studies, once-daily doses of 2.5 mg–10 mg were similar in their effect, lowering supine or standing systolic and diastolic blood pressures 24 hours after dosing by about 6/4 mmHg more than placebo. In comparisons of peak vs. trough effect, the trough effect represented about 50–60% of the peak response. In a titration study comparing divided (bid) vs. qd treatment, the divided regimen was superior, indicating that for some patients, the antihypertensive effect with once-daily dosing is not adequately maintained.
In most trials, the antihypertensive effect of ALTACE increased during the first several weeks of repeated measurements. The antihypertensive effect of ALTACE has been shown to continue during long-term therapy for at least 2 years. Abrupt withdrawal of ALTACE has not resulted in a rapid increase in blood pressure. ALTACE has been compared with other ACE inhibitors, beta-blockers, and thiazide diuretics. ALTACE was approximately as effective as other ACE inhibitors and as atenolol. In both Caucasians and Blacks, hydrochlorothiazide (25 or 50 mg) was significantly more effective than ramipril.
ALTACE was less effective in blacks than in Caucasians. The effectiveness of ALTACE was not influenced by age, sex, or weight.
In a baseline controlled study of 10 patients with mild essential hypertension, blood pressure reduction was accompanied by a 15% increase in renal blood flow. In healthy volunteers, glomerular filtration rate was unchanged.
14.2 Reduction in Risk of Myocardial Infarction, Stroke, and Death from Cardiovascular Causes
The HOPE study was a large, multicenter, randomized, double-blind, placebo-controlled, 2 × 2 factorial design study conducted in 9541 patients (4645 on ALTACE) who were 55 years or older and considered at high risk of developing a major cardiovascular event because of a history of coronary artery disease, stroke, peripheral vascular disease, or diabetes that was accompanied by at least one other cardiovascular risk factor (hypertension, elevated total cholesterol levels, low HDL levels, cigarette smoking, or documented microalbuminuria). Patients were either normotensive or under treatment with other antihypertensive agents. Patients were excluded if they had clinical heart failure or were known to have a low ejection fraction (<0.40). This study was designed to examine the long-term (mean of 5 years) effects of ALTACE (10 mg orally once daily) on the combined endpoint of myocardial infarction, stroke, or death from cardiovascular causes.
The HOPE study results showed that ALTACE (10 mg/day) significantly reduced the rate of myocardial infarction, stroke, or death from cardiovascular causes (826/4652 vs. 651/4645, relative risk 0.78), as well as the rates of the 3 components of the combined endpoint. The relative risk of the composite outcomes in the ALTACE group as compared to the placebo group was 0.78% (95% confidence interval, 0.70–0.86). The effect was evident after about 1 year of treatment.
| Outcome | Placebo (N=4652) n (%) | ALTACE (N=4645) n (%) | Relative Risk (95% CI) P-Value |
|---|---|---|---|
Combined Endpoint | |||
Myocardial infarction, stroke, or death from cardiovascular cause | 826 (17.8%) | 651 (14.0%) | 0.78 (0.70–0.86) |
Component Endpoint | |||
Death from cardiovascular causes | 377 (8.1%) | 282 (6.1%) | 0.74 (0.64–0.87) |
Myocardial infarction | 570 (12.3%) | 459 (9.9%) | 0.80 (0.70–0.90) |
Stroke | 226 (4.9%) | 156 (3.4%) | 0.68 (0.56–0.84) |
Overall Mortality | |||
Death from any cause | 569 (12.2%) | 482 (10.4%) | 0.84 (0.75–0.95) |
Figure 1. Kaplan-Meier Estimates of the Composite Outcome of Myocardial Infarction, Stroke, or Death from Cardiovascular Causes in the Ramipril Group and the Placebo Group
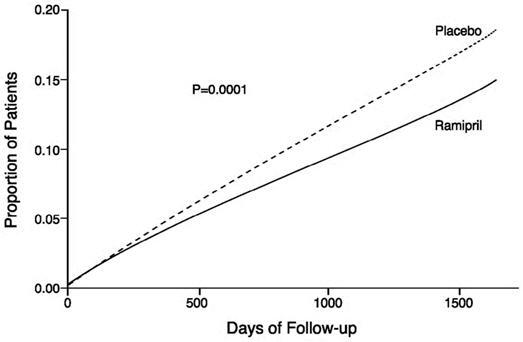
ALTACE was effective in different demographic subgroups (i.e., gender, age), subgroups defined by underlying disease (e.g., cardiovascular disease, hypertension), and subgroups defined by concomitant medication. There were insufficient data to determine whether or not ALTACE was equally effective in ethnic subgroups.
This study was designed with a prespecified substudy in diabetics with at least one other cardiovascular risk factor. Effects of ALTACE on the combined endpoint and its components were similar in diabetics (N=3577) to those in the overall study population.
| Outcome | Placebo (N=1769) n (%) | ALTACE (N=1808) n (%) | Relative Risk Reduction (95% CI) P-Value |
|---|---|---|---|
Combined Endpoint | |||
Myocardial infarction, stroke, or death from cardiovascular cause | 351 (19.8%) | 277 (15.3%) | 0.25 (0.12–0.36) |
Component Endpoint | |||
Death from cardiovascular causes | 172 (9.7%) | 112 (6.2%) | 0.37 (0.21–0.51) |
Myocardial infarction | 229 (12.9%) | 185 (10.2%) | 0.22 (0.06–0.36) |
Stroke | 108 (6.1%) | 76 (4.2%) | 0.33 (0.10–0.50) |
Figure 2. The Beneficial Effect of Treatment with ALTACE on the Composite Outcome of Myocardial Infarction, Stroke, or Death from Cardiovascular Causes Overall and in Various Subgroups
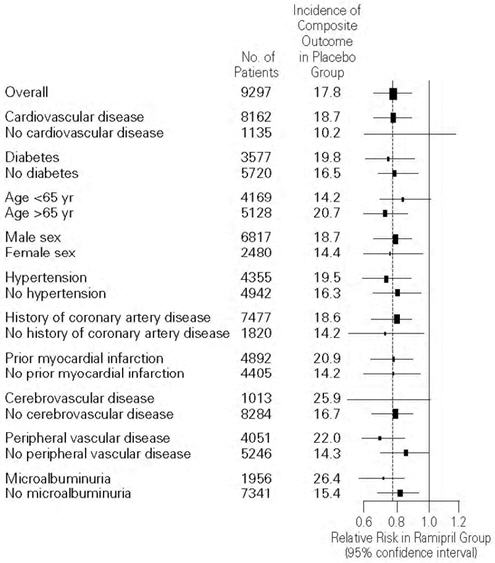
Cerebrovascular disease was defined as stroke or transient ischemic attacks. The size of each symbol is proportional to the number of patients in each group. The dashed line indicates overall relative risk.
The benefits of ALTACE were observed among patients who were taking aspirin or other anti-platelet agents, beta-blockers, and lipid-lowering agents as well as diuretics and calcium channel blockers.
14.3 Heart Failure Post-Myocardial Infarction
ALTACE was studied in the AIRE trial. This was a multinational (mainly European) 161-center, 2006-patient, double-blind, randomized, parallel-group study comparing ALTACE to placebo in stable patients, 2–9 days after an acute myocardial infarction, who had shown clinical signs of congestive heart failure at any time after the myocardial infarction. Patients in severe (NYHA class IV) heart failure, patients with unstable angina, patients with heart failure of congenital or valvular etiology, and patients with contraindications to ACE inhibitors were all excluded. The majority of patients had received thrombolytic therapy at the time of the index infarction, and the average time between infarction and initiation of treatment was 5 days.
Patients randomized to ALTACE treatment were given an initial dose of 2.5 mg twice daily. If the initial regimen caused undue hypotension, the dose was reduced to 1.25 mg, but in either event doses were titrated upward (as tolerated) to a target regimen (achieved in 77% of patients randomized to ALTACE) of 5 mg twice daily. Patients were then followed for an average of 15 months, with the range of follow-up between 6 and 46 months.
The use of ALTACE was associated with a 27% reduction (p=0.002) in the risk of death from any cause; about 90% of the deaths that occurred were cardiovascular, mainly sudden death. The risks of progression to severe heart failure and of congestive heart failure-related hospitalization were also reduced, by 23% (p=0.017) and 26% (p=0.011), respectively. The benefits of ALTACE therapy were seen in both genders, and they were not affected by the exact timing of the initiation of therapy, but older patients may have had a greater benefit than those under 65. The benefits were seen in patients on (and not on) various concomitant medications. At the time of randomization these included aspirin (about 80% of patients), diuretics (about 60%), organic nitrates (about 55%), beta-blockers (about 20%), calcium channel blockers (about 15%), and digoxin (about 12%).
How Supplied/Storage and Handling
16 HOW SUPPLIED/STORAGE AND HANDLING
ALTACE is available in 1.25 mg, 2.5 mg, 5 mg, and 10 mg hard gelatin capsules. Descriptions of ALTACE capsules are summarized below.
| Capsule Strength | Capsule Color | Package Configuration | NDC# |
|---|---|---|---|
1.25 mg | yellow | Bottle of 100 | 61570-110-01 |
2.5 mg | orange | Bottle of 100 | 61570-111-01 |
5 mg | red | Bottle of 100 | 61570-112-01 |
10 mg | Process Blue | Bottle of 100 | 61570-120-01 |
Patient Counseling Information
17 PATIENT COUNSELING INFORMATION
Angioedema
Angioedema, including laryngeal edema, can occur with treatment with ACE inhibitors, especially following the first dose. Advise patients to immediately report any signs or symptoms suggesting angioedema (swelling of face, eyes, lips, or tongue, or difficulty in breathing) and to temporarily discontinue drug until they have consulted with the prescribing physician.
Neutropenia
Advise patients to promptly report any indication of infection (e.g., sore throat, fever), which could be a sign of neutropenia.
Symptomatic Hypotension
Inform patients that light-headedness can occur, especially during the first days of therapy, and it should be reported. Advise patients to discontinue ALTACE if syncope (fainting) occurs, and to follow up with their health care providers.
Inform patients that inadequate fluid intake or excessive perspiration, diarrhea, or vomiting while taking ALTACE can lead to an excessive fall in blood pressure, with the same consequences of lightheadedness and possible syncope.
Other

Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Contact Medical Information. 9AM-5PM ET Monday to Friday; excluding holidays.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.