paclitaxel injection, USP Clinical Studies
CLINICAL STUDIES
Ovarian Carcinoma:
First-Line Data: The safety and efficacy of paclitaxel followed by cisplatin in patients with advanced ovarian cancer and no prior chemotherapy were evaluated in 2, Phase 3 multicenter, randomized, controlled trials. In an Intergroup study led by the European Organization for Research and Treatment of Cancer involving the Scandinavian Group NOCOVA, the National Cancer Institute of Canada, and the Scottish Group, 680 patients with Stage IIB–C, III, or IV disease (optimally or non-optimally debulked) received either paclitaxel 175 mg/m2 infused over 3 hours followed by cisplatin 75 mg/m2 (Tc) or cyclophosphamide 750 mg/m2 followed by cisplatin 75 mg/m2 (Cc) for a median of 6 courses. Although the protocol allowed further therapy, only 15% received both drugs for 9 or more courses. In a study conducted by the Gynecological Oncology Group (GOG), 410 patients with Stage III or IV disease (>1 cm residual disease after staging laparotomy or distant metastases) received either paclitaxel 135 mg/m2 infused over 24 hours followed by cisplatin 75 mg/m2 or cyclophosphamide 750 mg/m2 followed by cisplatin 75 mg/m2 for 6 courses.
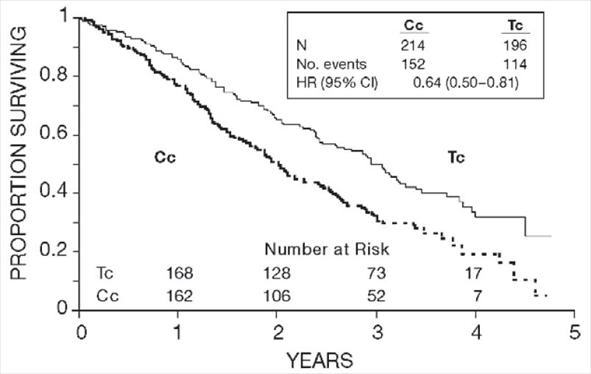
In both studies, patients treated with paclitaxel in combination with cisplatin had significantly higher response rate, longer time to progression, and longer survival time compared with standard therapy. These differences were also significant for the subset of patients in the Intergroup study with non-optimally debulked disease, although the study was not fully powered for subset analyses (Tables 2A and 2B). Kaplan- Meier survival curves for each study are shown in Figures 1 and 2.
Intergroup (non-optimally | GOG-111 | |||||
T175/3a | C750a | T135/24a | C750a | |||
• Clinical Responseb - rate (percent) - p-valuec | (n=153) 58 | 0.016 | (n=153) 43 | (n=113) 62 | 0.04 | (n=127) 48 |
• Time to Progression - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc |
13.2 | 0.0060 0.76 0.62–0.92 |
9.9 |
16.6 | 0.0008 0.70 0.56–0.86 |
13.0 |
• Survival - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc |
29.5 | 0.0057 73 0.58-0.91 |
21.9 |
35.5 | 0.0002 0.64 0.50-0.81 |
24.2 |
a Paclitaxel dose in mg/m2/infusion duration in hours; cyclophosphamide and cisplatin doses in mg/m2. b Among patients with measurable disease only. c Unstratified for the Intergroup Study, Stratified for Study GOG-111. | ||||||
T175/3a c75 (n=342) | C750a c75 (n=338) | ||
• Clinical Responseb - rate (percent) - p-valuec | (n=162) 59 | 0.014 | (n=161) 45 |
• Time to Progression - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc |
15.3 | 0.0005 0.74 0.63–0.88 |
11.5 |
• Survival - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc |
35.6 | 0.0016 0.73 0.60–0.89 |
25.9 |
a Paclitaxel dose in mg/m2/infusion duration in hours; cyclophosphamide and cisplatin doses in mg/m2. b Among patients with measurable disease only. c Unstratified. | |||
Figure 1. Survival: Cc Versus Tc (Intergroup)
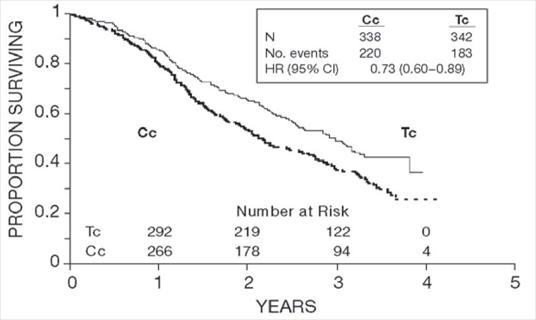
Figure 2. Survival: Cc Versus Tc (GOG-111)
The adverse event profile for patients receiving paclitaxel in combination with cisplatin in these studies was qualitatively consistent with that seen for the pooled analysis of data from 812 patients treated with single-agent paclitaxel in 10 clinical studies. These adverse events and adverse events from the Phase 3 first-line ovarian carcinoma studies are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 11) and narrative form.
Second-Line Data: Data from five Phase 1 & 2 clinical studies (189 patients), a multicenter randomized Phase 3 study (407 patients), as well as an interim analysis of data from more than 300 patients enrolled in a treatment referral center program were used in support of the use of paclitaxel in patients who have failed initial or subsequent chemotherapy for metastatic carcinoma of the ovary. Two of the Phase 2 studies (92 patients) utilized an initial dose of 135 to 170 mg/m2 in most patients (>90%) administered over 24 hours by continuous infusion. Response rates in these two studies were 22% (95% CI: 11% to 37%) and 30% (95% CI: 18% to 46%) with a total of 6 complete and 18 partial responses in 92 patients. The median duration of overall response in these two studies measured from the first day of treatment was 7.2 months (range: 3.5-15.8 months) and 7.5 months (range: 5.3-17.4 months), respectively. The median survival was 8.1 months (range: 0.2-36.7 months) and 15.9 months (range: 1.8-34.5+ months).
The Phase 3 study had a bifactorial design and compared the efficacy and safety of paclitaxel, administered at two different doses (135 or 175 mg/m2 and schedules (3- or 24-hour infusion). The overall response rate for the 407 patients was 16.2% (95% CI: 12.8% to 20.2%), with 6 complete and 60 partial responses. Duration of response, measured from the first day of treatment was 8.3 months (range: 3.2-21.6 months). Median time to progression was 3.7 months (range: 0.1+ - 25.1+ months). Median survival was 11.5 months (range: 0.2-26.3+ months).
Response rates, median survival, and median time to progression for the 4 arms are given in the following table.
175/3 (n=96) | 175/24 (n=106) | 135/3 (n=99) | 135/24 (n=106) | |
• Response - rate (percent) - 95% Confidence Interval | 14.6 (8.5-23.6) | 21.7 (14.5-31.0) | 15.2 (9.0-24.1) | 13.2 (7.7-21.5) |
• Time to Progression - median (months) - 95% Confidence Interval | 4.4 (3.0-5.6) | 4.2 (3.5-5.1) | 3.4 (2.8-4.2) | 2.8 (1.9-4.0) |
• Survival - median (months) - 95% Confidence Interval | 11.5 (8.4-14.4) | 11.8 (8.9-14.6) | 13.1 (9.1-14.6) | 10.7 (8.1-13.6) |
Analyses were performed as planned by the bifactorial study design described in the protocol, by comparing the two doses (135 or 175 mg/m2) irrespective of the schedule (3 or 24 hours) and the two schedules irrespective of dose. Patients receiving the 175 mg/m2 dose had a response rate similar to that for those receiving the 135 mg/m2 dose: 18% vs. 14% (p=0.28). No difference in response rate was detected when comparing the 3-hour with the 24-hour infusion: 15% vs. 17% (p=0.50). Patients receiving the 175 mg/m2 dose of paclitaxel had a longer time to progression than those receiving the 135 mg/m2 dose: median 4.2 vs. 3.1 months (p=0.03). The median time to progression for patients receiving the 3-hour vs. the 24-hour infusion was 4.0 months vs. 3.7 months, respectively. Median survival was 11.6 months in patients receiving the 175 mg/m2 dose of paclitaxel and 11.0 months in patients receiving the 135 mg/m2 dose (p=0.92). Median survival was 11.7 months for patients receiving the 3-hour infusion of paclitaxel and 11.2 months for patients receiving the 24 hour infusion (p=0.91). These statistical analyses should be viewed with caution because of the multiple comparisons made.
Paclitaxel remained active in patients who had developed resistance to platinum-containing therapy (defined as tumor progression while on, or tumor relapse within 6 months from completion of, a platinum-containing regimen) with response rates of 14% in the Phase 3 study and 31% in the Phase 1 & 2 clinical studies.
The adverse event profile in this Phase 3 study was consistent with that seen for the pooled analysis of data from 812 patients treated in 10 clinical studies. These adverse events and adverse events from the Phase 3 second-line ovarian carcinoma study are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 12) and narrative form.
The results of this randomized study support the use of Paclitaxel Injection, USP at doses of 135 to 175 mg/m2, administered by a 3-hour intravenous infusion. The same doses administered by 24-hour infusion were more toxic. However, the study had insufficient power to determine whether a particular dose and schedule produced superior efficacy.
Breast Carcinoma:
Adjuvant Therapy
A Phase 3 Intergroup study (Cancer and Leukemia Group B [CALGB], Eastern Cooperative Oncology Group [ECOG], North Central Cancer Treatment Group [NCCTG], and Southwest Oncology Group [SWOG]) randomized 3170 patients with node-positive breast carcinoma to adjuvant therapy with paclitaxel or to no further chemotherapy following 4 courses of doxorubicin and cyclophosphamide (AC). This multicenter trial was conducted in women with histologically positive lymph nodes following either a mastectomy or segmental mastectomy and nodal dissections. The 3 x 2 factorial study was designed to assess the efficacy and safety of 3 different dose levels of doxorubicin (A) and to evaluate the effect of the addition of paclitaxel administered following the completion of AC therapy. After stratification for the number of positive lymph nodes (1–3, 4–9, or 10+), patients were randomized to receive cyclophosphamide at a dose of 600 mg/m2 and doxorubicin at doses of either 60 mg/m2 (on day 1), 75 mg/m2 (in 2 divided doses on days 1 and 2), or 90 mg/m2 (in 2 divided doses on days 1 and 2 with prophylactic G-CSF support and ciprofloxacin) every 3 weeks for 4 courses and either paclitaxel 175 mg/m2 as a 3-hour infusion every 3 weeks for 4 additional courses or no additional chemotherapy. Patients whose tumors were positive were to receive subsequent tamoxifen treatment (20 mg daily for 5 years); patients who received segmental mastectomies prior to study were to receive breast irradiation after recovery from treatment-related toxicities.
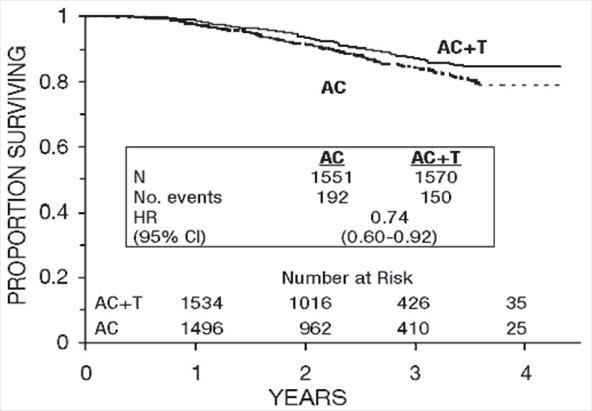
At the time of the current analysis, median follow-up was 30.1 months. Of the 2066 patients who were hormone receptor positive, 93% received tamoxifen. The primary analyses of disease-free survival and overall survival used multivariate Cox models, which included paclitaxel administration, doxorubicin dose, number of positive lymph nodes, tumor size, menopausal status, and estrogen receptor status as factors. Based on the model for disease-free survival, patients receiving AC followed by paclitaxel had a 22% reduction in the risk of disease recurrence compared to patients randomized to AC alone (Hazard Ratio [HR]=0.78, 95% CI, 0.67–0.91, p=0.0022). They also had a 26% reduction in the risk of death (HR=0.74, 95% CI, 0.60–0.92, p=0.0065). For disease-free survival and overall survival, p-values were not adjusted for interim analyses. Kaplan-Meier curves are shown in Figures 3 and 4. Increasing the dose of doxorubicin higher than 60 mg/m2 had no effect on either disease-free survival or overall survival.
Figure 3. Disease-Free Survival: AC Versus AC+T
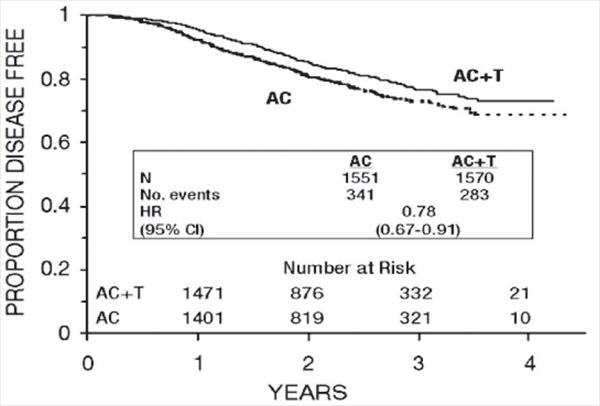
Figure 4. Survival: AC Versus AC+T
Subset analyses: Subsets defined by variables of known prognostic importance in adjuvant breast carcinoma were examined, including number of positive lymph nodes, tumor size, hormone receptor status, and menopausal status. Such analyses must be interpreted with care, as the most secure finding is the overall study result. In general, a reduction in hazard similar to the overall reduction was seen with paclitaxel for both disease-free and overall survival in all of the larger subsets with one exception; patients with receptor-positive tumors had a smaller reduction in hazard (HR=0.92) for disease-free survival with paclitaxel than other groups. Results of subset analyses are shown in Table 4.
Patient Subset |
| Disease-Free Survival | Overall Survival | ||
No. of Patients | No. of Recurrences | Hazard Ratio (95% CI) | No. of Deaths | Hazard Ratio (95% CI) | |
• No. of Positive Nodes 1-3
4-9
10+ |
1449
1310
360 |
221
274
129 |
0.72 (0.55-0.94) 0.78 (0.61-0.99) 0.93 (0.66-1.31) |
107
148
87 |
0.76 (0.52-1.12) 0.66 (0.47-0.91) 0.90 (0.59-1.36) |
• Tumor Size (cm) ≤2
>2 and ≤5
>5 |
1096
1611
397
|
153
358
111
|
0.79 (0.57-1.08) 0.79 (0.64-0.97) 0.75 (0.51-1.08) |
67
201
72 |
0.73 (0.45-1.18) 0.74 (0.56-0.98) 0.73 (0.46-1.16) |
• Menopausal Status Pre
Post |
1929
1183 |
374
250 |
0.83 (0.67-1.01) 0.73 (0.57-0.93) |
187
155 |
0.72 (0.54-0.97) 0.77 (0.56-1.06) |
• Receptor Status Positivea
Negative/Unknownb |
2066
1055 |
293
331 |
0.92 (0.73-1.16) 0.68 (0.55-0.85) |
126
216 |
0.83 (0.59-1.18) 0.71 (0.54-0.93) |
a Positive for either estrogen or progesterone receptors. b Negative or missing for both estrogen and progesterone receptors (both missing: n=15). | |||||
These retrospective subgroup analyses suggest that the beneficial effect of paclitaxel is clearly established in the receptor-negative subgroup, but the benefit in receptor-positive patients is not yet clear. With respect to menopausal status, the benefit of paclitaxel is consistent (see Table 4 and Figures 5–8).
Figure 5. Disease-Free Survival: Receptor Status Negative/Unknown AC Versus AC+T
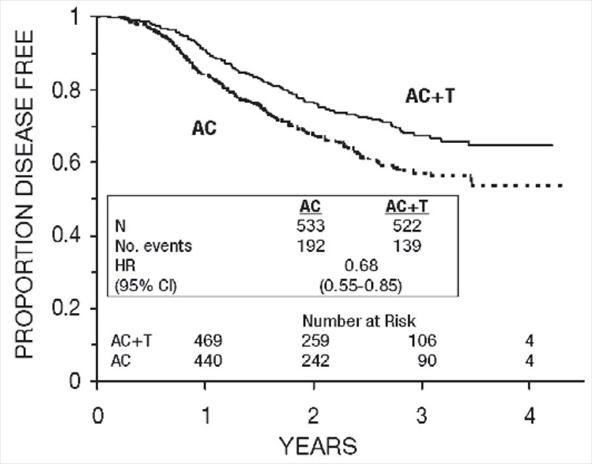
Figure 6. Disease-Free Survival: Receptor Status Positive AC Versus AC+T
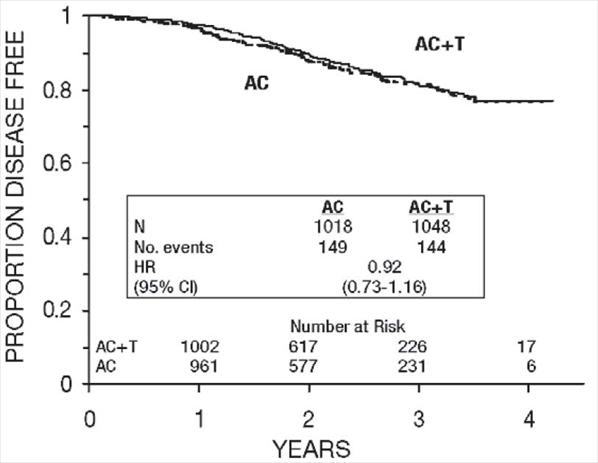
Figure 7. Disease-Free Survival: Premenopausal AC Versus AC+T
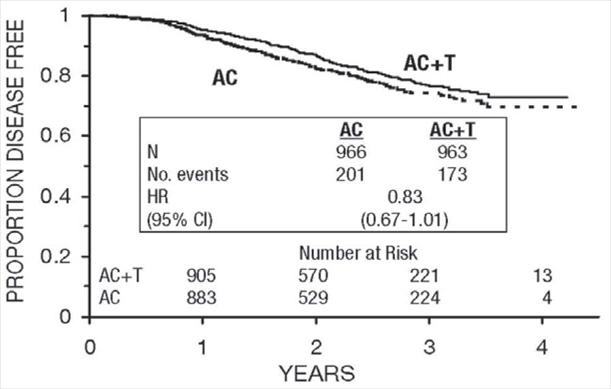
Figure 8. Disease-Free Survival: Postmenopausal AC Versus AC+T
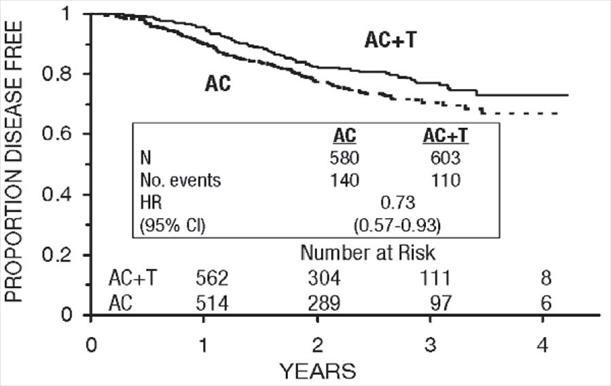
The adverse event profile for the patients who received paclitaxel subsequent to AC was consistent with that seen in the pooled analysis of data from 812 patients (Table 10) treated with single-agent paclitaxel in 10 clinical studies. These adverse events are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 13) and narrative form.
After Failure of Initial Chemotherapy: Data from 83 patients accrued in three Phase 2 open label studies and from 471 patients enrolled in a Phase 3 randomized study were available to support the use of paclitaxel in patients with metastatic breast carcinoma.
Phase 2 Open Label Studies: Two studies were conducted in 53 patients previously treated with a maximum of one prior chemotherapeutic regimen. Paclitaxel was administered in these two trials as a 24-hour infusion at initial doses of 250 mg/m2 (with G-CSF support) or 200 mg/m2. The response rates were 57% (95% CI: 37% to 75%) and 52% (95% CI: 32% to 72%), respectively. The third Phase 2 study was conducted in extensively pretreated patients who had failed anthracycline therapy and who had received a minimum of two chemotherapy regimens for the treatment of metastatic disease. The dose of paclitaxel was 200 mg/m2 as a 24-hour infusion with G-CSF support. Nine of 30 patients achieved a partial response, for a response rate of 30% (95% CI: 15% to 50%).
Phase 3 Randomized Study: This multicenter trial was conducted in patients previously treated with one or two regimens of chemotherapy. Patients were randomized to receive paclitaxel at a dose of either 175 mg/m2 or 135 mg/m2 given as a 3-hour infusion. In the 471 patients enrolled, 60% had symptomatic disease with impaired performance status at study entry, and 73% had visceral metastases. These patients had failed prior chemotherapy either in the adjuvant setting (30%), the metastatic setting (39%), or both (31%). Sixty-seven percent of the patients had been previously exposed to anthracyclines and 23% of them had disease considered resistant to this class of agents.
The overall response rate for the 454 evaluable patients was 26% (95% CI: 22% to 30%), with 17 complete and 99 partial responses. The median duration of response, measured from the first day of treatment, was 8.1 months (range: 3.4-18.1+ months). Overall for the 471 patients, the median time to progression was 3.5 months (range: 0.03-17.1 months). Median survival was 11.7 months (range: 0-18.9 months).
Response rates, median survival and median time to progression for the 2 arms are given in the following table.
| 175/3 (n=235) |
| 135/3 (n=236) |
• Response - rate (percent) - p-value | 28 | 0.135 | 22 |
• Time to Progression - median (months) - p-value | 4.2 | 0.027 | 3.0 |
• Survival - median (months) - p-value | 11.7 | 0.321 | 10.5 |
The adverse event profile of the patients who received single-agent Paclitaxel Injection, USP, in the Phase 3 study was consistent with that seen for the pooled analysis of data from 812 patients treated in 10 clinical studies. These adverse events and adverse events from the Phase 3 breast carcinoma study are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 14) and narrative form.
Non-Small Cell Lung Carcinoma (NSCLC)
In a Phase 3 open-label randomized study conducted by the ECOG, 599 patients were randomized to either paclitaxel (T) 135 mg/m2 as a 24-hour infusion in combination with cisplatin (c) 75 mg/m2, paclitaxel (T) 250 mg/m2 as a 24-hour infusion in combination with cisplatin (c) 75 mg/m2 with G-CSF support, or cisplatin (c) 75 mg/m2 on day 1, followed by etoposide (VP) 100 mg/m2 on days 1, 2, and 3 (control). Response rates, median time to progression, median survival, and 1-year survival rates are given in the following table. The reported p-values have not been adjusted for multiple comparisons. There were statistically significant differences favoring each of the paclitaxel plus cisplatin arms for response rate and time to tumor progression. There was no statistically significant difference in survival between either paclitaxel plus cisplatin arm and the cisplatin plus etoposide arm.
| T135/24 c75 (n=198) | T250/24 c75 (n=201) | VP100a c75 (n=200) |
• Response - rate (percent) - p-valueb | 25 0.001 | 23 <0.001 | 12 |
• Time to Progression - median (months) - p-valueb | 4.3 0.05 | 4.9 0.004 | 2.7 |
• Survival - median (months) - p-value b | 9.3 0.12 | 10.0 0.08 | 7.4 |
• 1-Year Survival - percent of patients | 36 | 40 | 32 |
a Etoposide (VP) 100 mg/m2 was administered IV on days 1, 2, and 3. b Compared to cisplatin/etoposide. | |||
In the ECOG study, the Functional Assessment of Cancer Therapy-Lung (FACT-L) questionnaire had 7 subscales that measured subjective assessment of treatment. Of the 7, the Lung Cancer Specific Symptoms subscale favored the paclitaxel 135 mg/m2/24 hour plus cisplatin arm compared to the cisplatin/etoposide arm. For all other factors, there was no difference in the treatment groups.
The adverse event profile for patients who received paclitaxel in combination with cisplatin in this study was generally consistent with that seen for the pooled analysis of data from 812 patients treated with single-agent paclitaxel in 10 clinical studies. These adverse events and adverse events from the Phase 3 first-line NSCLC study are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 15) and narrative form.
AIDS-Related Kaposi’s Sarcoma
Data from 2, Phase 2 open-label studies support the use of paclitaxel as second-line therapy in patients with AIDS-related Kaposi’s sarcoma. Fifty-nine of the 85 patients enrolled in these studies had previously received systemic therapy, including interferon alpha (32%), DaunoXome (31%), DOXIL (2%), and doxorubicin containing chemotherapy (42%), with 64% having received prior anthracyclines. Eighty-five percent of the pretreated patients had progressed on, or could not tolerate, prior systemic therapy.1
In Study CA139-174, patients received paclitaxel at 135 mg/m2 as a 3-hour infusion every 3 weeks (intended dose intensity 45 mg/m2/week). If no dose-limiting toxicity was observed, patients were to receive 155 mg/m2 and 175 mg/m2 in subsequent courses. Hematopoietic growth factors were not to be used initially. In Study CA139-281, patients received paclitaxel at 100 mg/m2 as a 3-hour infusion every 2 weeks (intended dose intensity 50 mg/m2/week). In this study patients could be receiving hematopoietic growth factors before the start of paclitaxel therapy, or this support was to be initiated as indicated; the dose of paclitaxel was not increased. The dose intensity of paclitaxel used in this patient population was lower than the dose intensity recommended for other solid tumors.
All patients had widespread and poor-risk disease. Applying the ACTG staging criteria to patients with prior systemic therapy, 93% were poor risk for extent of disease (T1), 88% had a CD4 count <200 cells/mm3 (I1), and 97% had poor risk considering their systemic illness (S1).
All patients in Study CA139-174 had a Karnofsky performance status of 80 or 90 at baseline; in Study CA139-281, there were 26 (46%) patients with a Karnofsky performance status of 70 or worse at baseline.
| Prior Systemic Therapy (n=59) |
Visceral ± edema ± oral ± cutaneous | 42 |
Edema or lymph nodes ± oral ± cutaneous | 41 |
Oral ± cutaneous | 10 |
Cutaneous only | 7 |
Although the planned dose intensity in the 2 studies was slightly different (45 mg/m2/week in Study CA139-174 and 50 mg/m2/week in Study CA139-281), delivered dose intensity was 38 to 39 mg/m2/week in both studies, with a similar range (20–24 to 51–61).
Efficacy: The efficacy of paclitaxel was evaluated by assessing cutaneous tumor response according to the amended ACTG criteria and by seeking evidence of clinical benefit in patients in 6 domains of symptoms and/or conditions that are commonly related to AIDS-related Kaposi’s sarcoma.
Cutaneous Tumor Response (Amended ACTG Criteria): The objective response rate was 59% (95% CI, 46–72%) (35 of 59 patients) in patients with prior systemic therapy. Cutaneous responses were primarily defined as flattening of more than 50% of previously raised lesions.
| Prior Systemic Therapy (n=59) |
Complete response | 3 |
Partial response | 56 |
Stable disease | 29 |
Progression | 8 |
Early death/toxicity | 3 |
The median time to response was 8.1 weeks and the median duration of response measured from the first day of treatment was 10.4 months (95% CI, 7.0–11.0 months) for the patients who had previously received systemic therapy. The median time to progression was 6.2 months (95% CI, 4.6–8.7 months).
Additional Clinical Benefit: Most data on patient benefit were assessed retrospectively (plans for such analyses were not included in the study protocols). Nonetheless, clinical descriptions and photographs indicated clear benefit in some patients, including instances of improved pulmonary function in patients with pulmonary involvement, improved ambulation, resolution of ulcers, and decreased analgesic requirements in patients with Kaposi’s sarcoma (KS) involving the feet and resolution of facial lesions and edema in patients with KS involving the face, extremities, and genitalia.
Safety: The adverse event profile of paclitaxel administered to patients with advanced HIV disease and poor-risk AIDS-related Kaposi’s sarcoma was generally similar to that seen in the pooled analysis of data from 812 patients with solid tumors. These adverse events and adverse events from the Phase 2 second-line Kaposi’s sarcoma studies are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 16) and narrative form. In this immunosuppressed patient population, however, a lower dose intensity of paclitaxel and supportive therapy including hematopoietic growth factors in patients with severe neutropenia are recommended. Patients with AIDS-related Kaposi’s sarcoma may have more severe hematologic toxicities than patients with solid tumors.
Find paclitaxel injection, USP medical information:
Find paclitaxel injection, USP medical information:
paclitaxel injection, USP Quick Finder
Health Professional Information
Clinical Studies
CLINICAL STUDIES
Ovarian Carcinoma:
First-Line Data: The safety and efficacy of paclitaxel followed by cisplatin in patients with advanced ovarian cancer and no prior chemotherapy were evaluated in 2, Phase 3 multicenter, randomized, controlled trials. In an Intergroup study led by the European Organization for Research and Treatment of Cancer involving the Scandinavian Group NOCOVA, the National Cancer Institute of Canada, and the Scottish Group, 680 patients with Stage IIB–C, III, or IV disease (optimally or non-optimally debulked) received either paclitaxel 175 mg/m2 infused over 3 hours followed by cisplatin 75 mg/m2 (Tc) or cyclophosphamide 750 mg/m2 followed by cisplatin 75 mg/m2 (Cc) for a median of 6 courses. Although the protocol allowed further therapy, only 15% received both drugs for 9 or more courses. In a study conducted by the Gynecological Oncology Group (GOG), 410 patients with Stage III or IV disease (>1 cm residual disease after staging laparotomy or distant metastases) received either paclitaxel 135 mg/m2 infused over 24 hours followed by cisplatin 75 mg/m2 or cyclophosphamide 750 mg/m2 followed by cisplatin 75 mg/m2 for 6 courses.
In both studies, patients treated with paclitaxel in combination with cisplatin had significantly higher response rate, longer time to progression, and longer survival time compared with standard therapy. These differences were also significant for the subset of patients in the Intergroup study with non-optimally debulked disease, although the study was not fully powered for subset analyses (Tables 2A and 2B). Kaplan- Meier survival curves for each study are shown in Figures 1 and 2.
Intergroup (non-optimally | GOG-111 | |||||
T175/3a | C750a | T135/24a | C750a | |||
• Clinical Responseb - rate (percent) - p-valuec | (n=153) 58 | 0.016 | (n=153) 43 | (n=113) 62 | 0.04 | (n=127) 48 |
• Time to Progression - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc |
13.2 | 0.0060 0.76 0.62–0.92 |
9.9 |
16.6 | 0.0008 0.70 0.56–0.86 |
13.0 |
• Survival - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc |
29.5 | 0.0057 73 0.58-0.91 |
21.9 |
35.5 | 0.0002 0.64 0.50-0.81 |
24.2 |
a Paclitaxel dose in mg/m2/infusion duration in hours; cyclophosphamide and cisplatin doses in mg/m2. b Among patients with measurable disease only. c Unstratified for the Intergroup Study, Stratified for Study GOG-111. | ||||||
T175/3a c75 (n=342) | C750a c75 (n=338) | ||
• Clinical Responseb - rate (percent) - p-valuec | (n=162) 59 | 0.014 | (n=161) 45 |
• Time to Progression - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc |
15.3 | 0.0005 0.74 0.63–0.88 |
11.5 |
• Survival - median (months) - p-valuec - hazard ratio (HR)c - 95% CIc |
35.6 | 0.0016 0.73 0.60–0.89 |
25.9 |
a Paclitaxel dose in mg/m2/infusion duration in hours; cyclophosphamide and cisplatin doses in mg/m2. b Among patients with measurable disease only. c Unstratified. | |||
Figure 1. Survival: Cc Versus Tc (Intergroup)
Figure 2. Survival: Cc Versus Tc (GOG-111)
The adverse event profile for patients receiving paclitaxel in combination with cisplatin in these studies was qualitatively consistent with that seen for the pooled analysis of data from 812 patients treated with single-agent paclitaxel in 10 clinical studies. These adverse events and adverse events from the Phase 3 first-line ovarian carcinoma studies are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 11) and narrative form.
Second-Line Data: Data from five Phase 1 & 2 clinical studies (189 patients), a multicenter randomized Phase 3 study (407 patients), as well as an interim analysis of data from more than 300 patients enrolled in a treatment referral center program were used in support of the use of paclitaxel in patients who have failed initial or subsequent chemotherapy for metastatic carcinoma of the ovary. Two of the Phase 2 studies (92 patients) utilized an initial dose of 135 to 170 mg/m2 in most patients (>90%) administered over 24 hours by continuous infusion. Response rates in these two studies were 22% (95% CI: 11% to 37%) and 30% (95% CI: 18% to 46%) with a total of 6 complete and 18 partial responses in 92 patients. The median duration of overall response in these two studies measured from the first day of treatment was 7.2 months (range: 3.5-15.8 months) and 7.5 months (range: 5.3-17.4 months), respectively. The median survival was 8.1 months (range: 0.2-36.7 months) and 15.9 months (range: 1.8-34.5+ months).
The Phase 3 study had a bifactorial design and compared the efficacy and safety of paclitaxel, administered at two different doses (135 or 175 mg/m2 and schedules (3- or 24-hour infusion). The overall response rate for the 407 patients was 16.2% (95% CI: 12.8% to 20.2%), with 6 complete and 60 partial responses. Duration of response, measured from the first day of treatment was 8.3 months (range: 3.2-21.6 months). Median time to progression was 3.7 months (range: 0.1+ - 25.1+ months). Median survival was 11.5 months (range: 0.2-26.3+ months).
Response rates, median survival, and median time to progression for the 4 arms are given in the following table.
175/3 (n=96) | 175/24 (n=106) | 135/3 (n=99) | 135/24 (n=106) | |
• Response - rate (percent) - 95% Confidence Interval | 14.6 (8.5-23.6) | 21.7 (14.5-31.0) | 15.2 (9.0-24.1) | 13.2 (7.7-21.5) |
• Time to Progression - median (months) - 95% Confidence Interval | 4.4 (3.0-5.6) | 4.2 (3.5-5.1) | 3.4 (2.8-4.2) | 2.8 (1.9-4.0) |
• Survival - median (months) - 95% Confidence Interval | 11.5 (8.4-14.4) | 11.8 (8.9-14.6) | 13.1 (9.1-14.6) | 10.7 (8.1-13.6) |
Analyses were performed as planned by the bifactorial study design described in the protocol, by comparing the two doses (135 or 175 mg/m2) irrespective of the schedule (3 or 24 hours) and the two schedules irrespective of dose. Patients receiving the 175 mg/m2 dose had a response rate similar to that for those receiving the 135 mg/m2 dose: 18% vs. 14% (p=0.28). No difference in response rate was detected when comparing the 3-hour with the 24-hour infusion: 15% vs. 17% (p=0.50). Patients receiving the 175 mg/m2 dose of paclitaxel had a longer time to progression than those receiving the 135 mg/m2 dose: median 4.2 vs. 3.1 months (p=0.03). The median time to progression for patients receiving the 3-hour vs. the 24-hour infusion was 4.0 months vs. 3.7 months, respectively. Median survival was 11.6 months in patients receiving the 175 mg/m2 dose of paclitaxel and 11.0 months in patients receiving the 135 mg/m2 dose (p=0.92). Median survival was 11.7 months for patients receiving the 3-hour infusion of paclitaxel and 11.2 months for patients receiving the 24 hour infusion (p=0.91). These statistical analyses should be viewed with caution because of the multiple comparisons made.
Paclitaxel remained active in patients who had developed resistance to platinum-containing therapy (defined as tumor progression while on, or tumor relapse within 6 months from completion of, a platinum-containing regimen) with response rates of 14% in the Phase 3 study and 31% in the Phase 1 & 2 clinical studies.
The adverse event profile in this Phase 3 study was consistent with that seen for the pooled analysis of data from 812 patients treated in 10 clinical studies. These adverse events and adverse events from the Phase 3 second-line ovarian carcinoma study are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 12) and narrative form.
The results of this randomized study support the use of Paclitaxel Injection, USP at doses of 135 to 175 mg/m2, administered by a 3-hour intravenous infusion. The same doses administered by 24-hour infusion were more toxic. However, the study had insufficient power to determine whether a particular dose and schedule produced superior efficacy.
Breast Carcinoma:
Adjuvant Therapy
A Phase 3 Intergroup study (Cancer and Leukemia Group B [CALGB], Eastern Cooperative Oncology Group [ECOG], North Central Cancer Treatment Group [NCCTG], and Southwest Oncology Group [SWOG]) randomized 3170 patients with node-positive breast carcinoma to adjuvant therapy with paclitaxel or to no further chemotherapy following 4 courses of doxorubicin and cyclophosphamide (AC). This multicenter trial was conducted in women with histologically positive lymph nodes following either a mastectomy or segmental mastectomy and nodal dissections. The 3 x 2 factorial study was designed to assess the efficacy and safety of 3 different dose levels of doxorubicin (A) and to evaluate the effect of the addition of paclitaxel administered following the completion of AC therapy. After stratification for the number of positive lymph nodes (1–3, 4–9, or 10+), patients were randomized to receive cyclophosphamide at a dose of 600 mg/m2 and doxorubicin at doses of either 60 mg/m2 (on day 1), 75 mg/m2 (in 2 divided doses on days 1 and 2), or 90 mg/m2 (in 2 divided doses on days 1 and 2 with prophylactic G-CSF support and ciprofloxacin) every 3 weeks for 4 courses and either paclitaxel 175 mg/m2 as a 3-hour infusion every 3 weeks for 4 additional courses or no additional chemotherapy. Patients whose tumors were positive were to receive subsequent tamoxifen treatment (20 mg daily for 5 years); patients who received segmental mastectomies prior to study were to receive breast irradiation after recovery from treatment-related toxicities.
At the time of the current analysis, median follow-up was 30.1 months. Of the 2066 patients who were hormone receptor positive, 93% received tamoxifen. The primary analyses of disease-free survival and overall survival used multivariate Cox models, which included paclitaxel administration, doxorubicin dose, number of positive lymph nodes, tumor size, menopausal status, and estrogen receptor status as factors. Based on the model for disease-free survival, patients receiving AC followed by paclitaxel had a 22% reduction in the risk of disease recurrence compared to patients randomized to AC alone (Hazard Ratio [HR]=0.78, 95% CI, 0.67–0.91, p=0.0022). They also had a 26% reduction in the risk of death (HR=0.74, 95% CI, 0.60–0.92, p=0.0065). For disease-free survival and overall survival, p-values were not adjusted for interim analyses. Kaplan-Meier curves are shown in Figures 3 and 4. Increasing the dose of doxorubicin higher than 60 mg/m2 had no effect on either disease-free survival or overall survival.
Figure 3. Disease-Free Survival: AC Versus AC+T
Figure 4. Survival: AC Versus AC+T
Subset analyses: Subsets defined by variables of known prognostic importance in adjuvant breast carcinoma were examined, including number of positive lymph nodes, tumor size, hormone receptor status, and menopausal status. Such analyses must be interpreted with care, as the most secure finding is the overall study result. In general, a reduction in hazard similar to the overall reduction was seen with paclitaxel for both disease-free and overall survival in all of the larger subsets with one exception; patients with receptor-positive tumors had a smaller reduction in hazard (HR=0.92) for disease-free survival with paclitaxel than other groups. Results of subset analyses are shown in Table 4.
Patient Subset |
| Disease-Free Survival | Overall Survival | ||
No. of Patients | No. of Recurrences | Hazard Ratio (95% CI) | No. of Deaths | Hazard Ratio (95% CI) | |
• No. of Positive Nodes 1-3
4-9
10+ |
1449
1310
360 |
221
274
129 |
0.72 (0.55-0.94) 0.78 (0.61-0.99) 0.93 (0.66-1.31) |
107
148
87 |
0.76 (0.52-1.12) 0.66 (0.47-0.91) 0.90 (0.59-1.36) |
• Tumor Size (cm) ≤2
>2 and ≤5
>5 |
1096
1611
397
|
153
358
111
|
0.79 (0.57-1.08) 0.79 (0.64-0.97) 0.75 (0.51-1.08) |
67
201
72 |
0.73 (0.45-1.18) 0.74 (0.56-0.98) 0.73 (0.46-1.16) |
• Menopausal Status Pre
Post |
1929
1183 |
374
250 |
0.83 (0.67-1.01) 0.73 (0.57-0.93) |
187
155 |
0.72 (0.54-0.97) 0.77 (0.56-1.06) |
• Receptor Status Positivea
Negative/Unknownb |
2066
1055 |
293
331 |
0.92 (0.73-1.16) 0.68 (0.55-0.85) |
126
216 |
0.83 (0.59-1.18) 0.71 (0.54-0.93) |
a Positive for either estrogen or progesterone receptors. b Negative or missing for both estrogen and progesterone receptors (both missing: n=15). | |||||
These retrospective subgroup analyses suggest that the beneficial effect of paclitaxel is clearly established in the receptor-negative subgroup, but the benefit in receptor-positive patients is not yet clear. With respect to menopausal status, the benefit of paclitaxel is consistent (see Table 4 and Figures 5–8).
Figure 5. Disease-Free Survival: Receptor Status Negative/Unknown AC Versus AC+T
Figure 6. Disease-Free Survival: Receptor Status Positive AC Versus AC+T
Figure 7. Disease-Free Survival: Premenopausal AC Versus AC+T
Figure 8. Disease-Free Survival: Postmenopausal AC Versus AC+T
The adverse event profile for the patients who received paclitaxel subsequent to AC was consistent with that seen in the pooled analysis of data from 812 patients (Table 10) treated with single-agent paclitaxel in 10 clinical studies. These adverse events are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 13) and narrative form.
After Failure of Initial Chemotherapy: Data from 83 patients accrued in three Phase 2 open label studies and from 471 patients enrolled in a Phase 3 randomized study were available to support the use of paclitaxel in patients with metastatic breast carcinoma.
Phase 2 Open Label Studies: Two studies were conducted in 53 patients previously treated with a maximum of one prior chemotherapeutic regimen. Paclitaxel was administered in these two trials as a 24-hour infusion at initial doses of 250 mg/m2 (with G-CSF support) or 200 mg/m2. The response rates were 57% (95% CI: 37% to 75%) and 52% (95% CI: 32% to 72%), respectively. The third Phase 2 study was conducted in extensively pretreated patients who had failed anthracycline therapy and who had received a minimum of two chemotherapy regimens for the treatment of metastatic disease. The dose of paclitaxel was 200 mg/m2 as a 24-hour infusion with G-CSF support. Nine of 30 patients achieved a partial response, for a response rate of 30% (95% CI: 15% to 50%).
Phase 3 Randomized Study: This multicenter trial was conducted in patients previously treated with one or two regimens of chemotherapy. Patients were randomized to receive paclitaxel at a dose of either 175 mg/m2 or 135 mg/m2 given as a 3-hour infusion. In the 471 patients enrolled, 60% had symptomatic disease with impaired performance status at study entry, and 73% had visceral metastases. These patients had failed prior chemotherapy either in the adjuvant setting (30%), the metastatic setting (39%), or both (31%). Sixty-seven percent of the patients had been previously exposed to anthracyclines and 23% of them had disease considered resistant to this class of agents.
The overall response rate for the 454 evaluable patients was 26% (95% CI: 22% to 30%), with 17 complete and 99 partial responses. The median duration of response, measured from the first day of treatment, was 8.1 months (range: 3.4-18.1+ months). Overall for the 471 patients, the median time to progression was 3.5 months (range: 0.03-17.1 months). Median survival was 11.7 months (range: 0-18.9 months).
Response rates, median survival and median time to progression for the 2 arms are given in the following table.
| 175/3 (n=235) |
| 135/3 (n=236) |
• Response - rate (percent) - p-value | 28 | 0.135 | 22 |
• Time to Progression - median (months) - p-value | 4.2 | 0.027 | 3.0 |
• Survival - median (months) - p-value | 11.7 | 0.321 | 10.5 |
The adverse event profile of the patients who received single-agent Paclitaxel Injection, USP, in the Phase 3 study was consistent with that seen for the pooled analysis of data from 812 patients treated in 10 clinical studies. These adverse events and adverse events from the Phase 3 breast carcinoma study are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 14) and narrative form.
Non-Small Cell Lung Carcinoma (NSCLC)
In a Phase 3 open-label randomized study conducted by the ECOG, 599 patients were randomized to either paclitaxel (T) 135 mg/m2 as a 24-hour infusion in combination with cisplatin (c) 75 mg/m2, paclitaxel (T) 250 mg/m2 as a 24-hour infusion in combination with cisplatin (c) 75 mg/m2 with G-CSF support, or cisplatin (c) 75 mg/m2 on day 1, followed by etoposide (VP) 100 mg/m2 on days 1, 2, and 3 (control). Response rates, median time to progression, median survival, and 1-year survival rates are given in the following table. The reported p-values have not been adjusted for multiple comparisons. There were statistically significant differences favoring each of the paclitaxel plus cisplatin arms for response rate and time to tumor progression. There was no statistically significant difference in survival between either paclitaxel plus cisplatin arm and the cisplatin plus etoposide arm.
| T135/24 c75 (n=198) | T250/24 c75 (n=201) | VP100a c75 (n=200) |
• Response - rate (percent) - p-valueb | 25 0.001 | 23 <0.001 | 12 |
• Time to Progression - median (months) - p-valueb | 4.3 0.05 | 4.9 0.004 | 2.7 |
• Survival - median (months) - p-value b | 9.3 0.12 | 10.0 0.08 | 7.4 |
• 1-Year Survival - percent of patients | 36 | 40 | 32 |
a Etoposide (VP) 100 mg/m2 was administered IV on days 1, 2, and 3. b Compared to cisplatin/etoposide. | |||
In the ECOG study, the Functional Assessment of Cancer Therapy-Lung (FACT-L) questionnaire had 7 subscales that measured subjective assessment of treatment. Of the 7, the Lung Cancer Specific Symptoms subscale favored the paclitaxel 135 mg/m2/24 hour plus cisplatin arm compared to the cisplatin/etoposide arm. For all other factors, there was no difference in the treatment groups.
The adverse event profile for patients who received paclitaxel in combination with cisplatin in this study was generally consistent with that seen for the pooled analysis of data from 812 patients treated with single-agent paclitaxel in 10 clinical studies. These adverse events and adverse events from the Phase 3 first-line NSCLC study are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 15) and narrative form.
AIDS-Related Kaposi’s Sarcoma
Data from 2, Phase 2 open-label studies support the use of paclitaxel as second-line therapy in patients with AIDS-related Kaposi’s sarcoma. Fifty-nine of the 85 patients enrolled in these studies had previously received systemic therapy, including interferon alpha (32%), DaunoXome (31%), DOXIL (2%), and doxorubicin containing chemotherapy (42%), with 64% having received prior anthracyclines. Eighty-five percent of the pretreated patients had progressed on, or could not tolerate, prior systemic therapy.1
In Study CA139-174, patients received paclitaxel at 135 mg/m2 as a 3-hour infusion every 3 weeks (intended dose intensity 45 mg/m2/week). If no dose-limiting toxicity was observed, patients were to receive 155 mg/m2 and 175 mg/m2 in subsequent courses. Hematopoietic growth factors were not to be used initially. In Study CA139-281, patients received paclitaxel at 100 mg/m2 as a 3-hour infusion every 2 weeks (intended dose intensity 50 mg/m2/week). In this study patients could be receiving hematopoietic growth factors before the start of paclitaxel therapy, or this support was to be initiated as indicated; the dose of paclitaxel was not increased. The dose intensity of paclitaxel used in this patient population was lower than the dose intensity recommended for other solid tumors.
All patients had widespread and poor-risk disease. Applying the ACTG staging criteria to patients with prior systemic therapy, 93% were poor risk for extent of disease (T1), 88% had a CD4 count <200 cells/mm3 (I1), and 97% had poor risk considering their systemic illness (S1).
All patients in Study CA139-174 had a Karnofsky performance status of 80 or 90 at baseline; in Study CA139-281, there were 26 (46%) patients with a Karnofsky performance status of 70 or worse at baseline.
| Prior Systemic Therapy (n=59) |
Visceral ± edema ± oral ± cutaneous | 42 |
Edema or lymph nodes ± oral ± cutaneous | 41 |
Oral ± cutaneous | 10 |
Cutaneous only | 7 |
Although the planned dose intensity in the 2 studies was slightly different (45 mg/m2/week in Study CA139-174 and 50 mg/m2/week in Study CA139-281), delivered dose intensity was 38 to 39 mg/m2/week in both studies, with a similar range (20–24 to 51–61).
Efficacy: The efficacy of paclitaxel was evaluated by assessing cutaneous tumor response according to the amended ACTG criteria and by seeking evidence of clinical benefit in patients in 6 domains of symptoms and/or conditions that are commonly related to AIDS-related Kaposi’s sarcoma.
Cutaneous Tumor Response (Amended ACTG Criteria): The objective response rate was 59% (95% CI, 46–72%) (35 of 59 patients) in patients with prior systemic therapy. Cutaneous responses were primarily defined as flattening of more than 50% of previously raised lesions.
| Prior Systemic Therapy (n=59) |
Complete response | 3 |
Partial response | 56 |
Stable disease | 29 |
Progression | 8 |
Early death/toxicity | 3 |
The median time to response was 8.1 weeks and the median duration of response measured from the first day of treatment was 10.4 months (95% CI, 7.0–11.0 months) for the patients who had previously received systemic therapy. The median time to progression was 6.2 months (95% CI, 4.6–8.7 months).
Additional Clinical Benefit: Most data on patient benefit were assessed retrospectively (plans for such analyses were not included in the study protocols). Nonetheless, clinical descriptions and photographs indicated clear benefit in some patients, including instances of improved pulmonary function in patients with pulmonary involvement, improved ambulation, resolution of ulcers, and decreased analgesic requirements in patients with Kaposi’s sarcoma (KS) involving the feet and resolution of facial lesions and edema in patients with KS involving the face, extremities, and genitalia.
Safety: The adverse event profile of paclitaxel administered to patients with advanced HIV disease and poor-risk AIDS-related Kaposi’s sarcoma was generally similar to that seen in the pooled analysis of data from 812 patients with solid tumors. These adverse events and adverse events from the Phase 2 second-line Kaposi’s sarcoma studies are described in the ADVERSE REACTIONS section in tabular (Tables 10 and 16) and narrative form. In this immunosuppressed patient population, however, a lower dose intensity of paclitaxel and supportive therapy including hematopoietic growth factors in patients with severe neutropenia are recommended. Patients with AIDS-related Kaposi’s sarcoma may have more severe hematologic toxicities than patients with solid tumors.
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.