INLYTA® Clinical Pharmacology
(axitinib)
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Axitinib has been shown to inhibit receptor tyrosine kinases including vascular endothelial growth factor receptors (VEGFR)-1, VEGFR-2, and VEGFR-3 at therapeutic plasma concentrations. These receptors are implicated in pathologic angiogenesis, tumor growth, and cancer progression. VEGF-mediated endothelial cell proliferation and survival were inhibited by axitinib in vitro and in mouse models. Axitinib was shown to inhibit tumor growth and phosphorylation of VEGFR-2 in tumor xenograft mouse models.
12.2 Pharmacodynamics
The effect of a single oral dose of INLYTA (5 mg) in the absence and presence of 400 mg ketoconazole on the QTc interval was evaluated in a randomized, single-blinded, two-way crossover study in 35 healthy subjects. No large changes in mean QTc interval (i.e., >20 ms) from placebo were detected up to 3 hours post-dose. However, small increases in mean QTc interval (i.e., <10 ms) cannot be ruled out.
12.3 Pharmacokinetics
The population pharmacokinetic analysis pooled data from 17 trials in healthy subjects and patients with cancer. A two-compartment disposition model with first-order absorption and lag-time adequately describes the axitinib concentration-time profile.
Absorption and Distribution
Following single oral 5-mg dose administration, the median Tmax ranged from 2.5 to 4.1 hours. Based on the plasma half-life, steady state is expected within 2 to 3 days of dosing. Dosing of axitinib at 5 mg twice daily resulted in approximately 1.4-fold accumulation compared to administration of a single dose. At steady state, axitinib exhibits approximately linear pharmacokinetics within the 1-mg to 20-mg dose range. The mean absolute bioavailability of axitinib after an oral 5 mg dose is 58%.
Compared to overnight fasting, administration of INLYTA with a moderate fat meal resulted in 10% lower AUC and a high fat, high-calorie meal resulted in 19% higher AUC. INLYTA can be administered with or without food [see Dosage and Administration (2.1)].
Axitinib is highly bound (>99%) to human plasma proteins with preferential binding to albumin and moderate binding to α1-acid glycoprotein. In patients with advanced RCC (n=20), at the 5 mg twice daily dose in the fed state, the geometric mean (CV%) Cmax and AUC0–24 were 27.8 (79%) ng/mL and 265 (77%) ng.h/mL, respectively. The geometric mean (CV%) clearance and apparent volume of distribution were 38 (80%) L/h and 160 (105%) L, respectively.
Metabolism and Elimination
The plasma half-life of INLYTA ranges from 2.5 to 6.1 hours. Axitinib is metabolized primarily in the liver by CYP3A4/5 and to a lesser extent by CYP1A2, CYP2C19, and UGT1A1. Following oral administration of a 5-mg radioactive dose of axitinib, approximately 41% of the radioactivity was recovered in feces and approximately 23% was recovered in urine. Unchanged axitinib, accounting for 12% of the dose, was the major component identified in feces. Unchanged axitinib was not detected in urine; the carboxylic acid and sulfoxide metabolites accounted for the majority of radioactivity in urine. In plasma, the N-glucuronide metabolite represented the predominant radioactive component (50% of circulating radioactivity) and unchanged axitinib and the sulfoxide metabolite each accounted for approximately 20% of the circulating radioactivity.
The sulfoxide and N-glucuronide metabolites show approximately ≥400-fold less in vitro potency against VEGFR-2 compared to axitinib.
Drug-Drug Interactions
Effects of Other Drugs on INLYTA
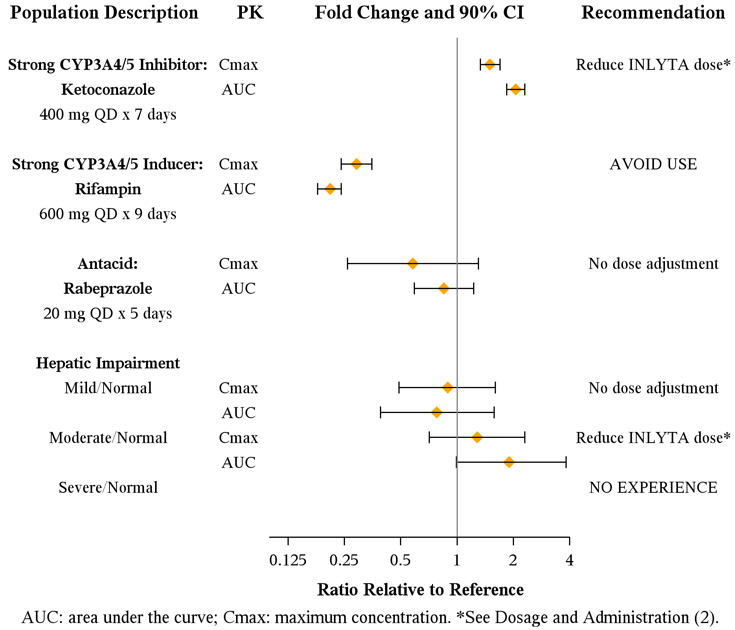
Axitinib is metabolized primarily in the liver by CYP3A4/5. Additionally, the aqueous solubility of axitinib is pH dependent, with higher pH resulting in lower solubility. The effects of a strong CYP3A4/5 inhibitor, a strong CYP3A4/5 inducer, and an antacid on the pharmacokinetics of axitinib are presented in Figure 1 [see Dosage and Administration (2.2) and Drug Interactions (7.1, 7.2)].
Figure 1. Impact of Co-administered Drugs and Hepatic Impairment on Axitinib Pharmacokinetics
Effects of INLYTA on Other Drugs
In vitro studies demonstrated that axitinib has the potential to inhibit CYP1A2 and CYP2C8. However, co-administration of axitinib with paclitaxel, a CYP2C8 substrate, did not increase plasma concentrations of paclitaxel in patients.
In vitro studies indicated that axitinib does not inhibit CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, or UGT1A1 at therapeutic plasma concentrations. In vitro studies in human hepatocytes indicated that axitinib does not induce CYP1A1, CYP1A2, or CYP3A4/5.
Axitinib is an inhibitor of the efflux transporter P-glycoprotein (P-gp) in vitro. However, INLYTA is not expected to inhibit P-gp at therapeutic plasma concentrations.
Specific Populations
Patients with Hepatic Impairment
The effects of hepatic impairment on the pharmacokinetics of axitinib are presented in Figure 1 [see Dosage and Administration (2.2), Warnings and Precautions (5.12), Use in Specific Populations (8.6)].
Patients with Renal Impairment
Population pharmacokinetic analysis (based on pre-existing renal function) was carried out in 590 healthy volunteers and patients, including five with severe renal impairment (15 mL/min ≤CLcr <29 mL/min), 64 with moderate renal impairment (30 mL/min ≤CLcr <59 mL/min), and 139 with mild renal impairment (60 mL/min ≤CLcr <89 mL/min). Mild to severe renal impairment did not have meaningful effects on the pharmacokinetics of axitinib. Data from only one patient with end-stage renal disease are available [see Use in Specific Populations (8.7)].
Other Intrinsic Factors
Population pharmacokinetic analyses indicate that there are no clinically relevant effects of age, gender, race, body weight, body surface area, UGT1A1 genotype, or CYP2C19 genotype on the clearance of axitinib.
INLYTA in Combination with Avelumab
When INLYTA 5 mg was administered in combination with avelumab 10 mg/kg, the respective exposures of INLYTA and avelumab were comparable to the single agents. There was no evidence to suggest a clinically relevant change of avelumab clearance over time in patients with advanced RCC.
Find INLYTA® medical information:
Find INLYTA® medical information:
INLYTA® Quick Finder
Health Professional Information
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Axitinib has been shown to inhibit receptor tyrosine kinases including vascular endothelial growth factor receptors (VEGFR)-1, VEGFR-2, and VEGFR-3 at therapeutic plasma concentrations. These receptors are implicated in pathologic angiogenesis, tumor growth, and cancer progression. VEGF-mediated endothelial cell proliferation and survival were inhibited by axitinib in vitro and in mouse models. Axitinib was shown to inhibit tumor growth and phosphorylation of VEGFR-2 in tumor xenograft mouse models.
12.2 Pharmacodynamics
The effect of a single oral dose of INLYTA (5 mg) in the absence and presence of 400 mg ketoconazole on the QTc interval was evaluated in a randomized, single-blinded, two-way crossover study in 35 healthy subjects. No large changes in mean QTc interval (i.e., >20 ms) from placebo were detected up to 3 hours post-dose. However, small increases in mean QTc interval (i.e., <10 ms) cannot be ruled out.
12.3 Pharmacokinetics
The population pharmacokinetic analysis pooled data from 17 trials in healthy subjects and patients with cancer. A two-compartment disposition model with first-order absorption and lag-time adequately describes the axitinib concentration-time profile.
Absorption and Distribution
Following single oral 5-mg dose administration, the median Tmax ranged from 2.5 to 4.1 hours. Based on the plasma half-life, steady state is expected within 2 to 3 days of dosing. Dosing of axitinib at 5 mg twice daily resulted in approximately 1.4-fold accumulation compared to administration of a single dose. At steady state, axitinib exhibits approximately linear pharmacokinetics within the 1-mg to 20-mg dose range. The mean absolute bioavailability of axitinib after an oral 5 mg dose is 58%.
Compared to overnight fasting, administration of INLYTA with a moderate fat meal resulted in 10% lower AUC and a high fat, high-calorie meal resulted in 19% higher AUC. INLYTA can be administered with or without food [see Dosage and Administration (2.1)].
Axitinib is highly bound (>99%) to human plasma proteins with preferential binding to albumin and moderate binding to α1-acid glycoprotein. In patients with advanced RCC (n=20), at the 5 mg twice daily dose in the fed state, the geometric mean (CV%) Cmax and AUC0–24 were 27.8 (79%) ng/mL and 265 (77%) ng.h/mL, respectively. The geometric mean (CV%) clearance and apparent volume of distribution were 38 (80%) L/h and 160 (105%) L, respectively.
Metabolism and Elimination
The plasma half-life of INLYTA ranges from 2.5 to 6.1 hours. Axitinib is metabolized primarily in the liver by CYP3A4/5 and to a lesser extent by CYP1A2, CYP2C19, and UGT1A1. Following oral administration of a 5-mg radioactive dose of axitinib, approximately 41% of the radioactivity was recovered in feces and approximately 23% was recovered in urine. Unchanged axitinib, accounting for 12% of the dose, was the major component identified in feces. Unchanged axitinib was not detected in urine; the carboxylic acid and sulfoxide metabolites accounted for the majority of radioactivity in urine. In plasma, the N-glucuronide metabolite represented the predominant radioactive component (50% of circulating radioactivity) and unchanged axitinib and the sulfoxide metabolite each accounted for approximately 20% of the circulating radioactivity.
The sulfoxide and N-glucuronide metabolites show approximately ≥400-fold less in vitro potency against VEGFR-2 compared to axitinib.
Drug-Drug Interactions
Effects of Other Drugs on INLYTA
Axitinib is metabolized primarily in the liver by CYP3A4/5. Additionally, the aqueous solubility of axitinib is pH dependent, with higher pH resulting in lower solubility. The effects of a strong CYP3A4/5 inhibitor, a strong CYP3A4/5 inducer, and an antacid on the pharmacokinetics of axitinib are presented in Figure 1 [see Dosage and Administration (2.2) and Drug Interactions (7.1, 7.2)].
Figure 1. Impact of Co-administered Drugs and Hepatic Impairment on Axitinib Pharmacokinetics
Effects of INLYTA on Other Drugs
In vitro studies demonstrated that axitinib has the potential to inhibit CYP1A2 and CYP2C8. However, co-administration of axitinib with paclitaxel, a CYP2C8 substrate, did not increase plasma concentrations of paclitaxel in patients.
In vitro studies indicated that axitinib does not inhibit CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, or UGT1A1 at therapeutic plasma concentrations. In vitro studies in human hepatocytes indicated that axitinib does not induce CYP1A1, CYP1A2, or CYP3A4/5.
Axitinib is an inhibitor of the efflux transporter P-glycoprotein (P-gp) in vitro. However, INLYTA is not expected to inhibit P-gp at therapeutic plasma concentrations.
Specific Populations
Patients with Hepatic Impairment
The effects of hepatic impairment on the pharmacokinetics of axitinib are presented in Figure 1 [see Dosage and Administration (2.2), Warnings and Precautions (5.12), Use in Specific Populations (8.6)].
Patients with Renal Impairment
Population pharmacokinetic analysis (based on pre-existing renal function) was carried out in 590 healthy volunteers and patients, including five with severe renal impairment (15 mL/min ≤CLcr <29 mL/min), 64 with moderate renal impairment (30 mL/min ≤CLcr <59 mL/min), and 139 with mild renal impairment (60 mL/min ≤CLcr <89 mL/min). Mild to severe renal impairment did not have meaningful effects on the pharmacokinetics of axitinib. Data from only one patient with end-stage renal disease are available [see Use in Specific Populations (8.7)].
Other Intrinsic Factors
Population pharmacokinetic analyses indicate that there are no clinically relevant effects of age, gender, race, body weight, body surface area, UGT1A1 genotype, or CYP2C19 genotype on the clearance of axitinib.
INLYTA in Combination with Avelumab
When INLYTA 5 mg was administered in combination with avelumab 10 mg/kg, the respective exposures of INLYTA and avelumab were comparable to the single agents. There was no evidence to suggest a clinically relevant change of avelumab clearance over time in patients with advanced RCC.
Health Professional Information
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.