TIKOSYN® Clinical Studies
(dofetilide)
CLINICAL STUDIES
Chronic Atrial Fibrillation and/or Atrial Flutter
Two randomized, parallel, double-blind, placebo-controlled, dose-response trials evaluated the ability of TIKOSYN 1) to convert patients with atrial fibrillation or atrial flutter (AF/AFl) of more than 1 week duration to normal sinus rhythm (NSR) and 2) to maintain NSR (delay time to recurrence of AF/AFl) after drug-induced or electrical cardioversion. A total of 996 patients with a one week to two year history of atrial fibrillation/atrial flutter were enrolled. Both studies randomized patients to placebo or to doses of TIKOSYN 125 mcg, 250 mcg, 500 mcg, or in one study a comparator drug, given twice a day (these doses were lowered based on calculated creatinine clearance and, in one of the studies, for QT interval or QTc). All patients were started on therapy in a hospital where their ECG was monitored (see DOSAGE AND ADMINISTRATION).
Patients were excluded from participation if they had had syncope within the past 6 months, AV block greater than first degree, MI or unstable angina within 1 month, cardiac surgery within 2 months, history of QT interval prolongation or polymorphic ventricular tachycardia associated with use of antiarrhythmic drugs, QT interval or QTc >440 msec, serum creatinine >2.5 mg/mL, significant diseases of other organ systems; used cimetidine; or used drugs known to prolong the QT interval.
Both studies enrolled mostly Caucasians (over 90%), males (over 70%), and patients ≥65 years of age (over 50%). Most (>90%) were NYHA Functional Class I or II. Approximately one-half had structural heart disease (including ischemic heart disease, cardiomyopathies, and valvular disease) and about one-half were hypertensive. A substantial proportion of patients were on concomitant therapy, including digoxin (over 60%), diuretics (over 20%), and ACE inhibitors (over 30%). About 90% were on anticoagulants.
Acute conversion rates are shown in Table 1 for randomized doses (doses were adjusted for calculated creatinine clearance and, in Study 1, for QT interval or QTc). Of patients who converted pharmacologically, approximately 70% converted within 24–36 hours.
| TIKOSYN Dose | Placebo | |||
|---|---|---|---|---|
| 125 mcg BID | 250 mcg BID | 500 mcg BID | ||
Study 1 | 5/82(6%) | 8/82(10%) | 23/77(30%) | 1/84(1%) |
Study 2 | 8/135(6%) | 14/133(11%) | 38/129(29%) | 2/137(1%) |
Patients who did not convert to NSR with randomized therapy within 48–72 hours had electrical cardioversion. Those patients remaining in NSR after conversion in hospital were continued on randomized therapy as outpatients (maintenance period) for up to one year unless they experienced a recurrence of atrial fibrillation/atrial flutter or withdrew for other reasons.
Table 2 shows, by randomized dose, the percentage of patients at 6 and 12 months in both studies who remained on treatment in NSR and the percentage of patients who withdrew because of recurrence of AF/AFl or adverse events.
| TIKOSYN Dose | Placebo | |||
|---|---|---|---|---|
| 125 mcg BID | 250 mcg BID | 500 mcg BID | ||
| Note that columns do not add up to 100% due to discontinuations for "other" reasons. | ||||
Study 1 | ||||
Randomized | 82 | 82 | 77 | 84 |
Achieved NSR | 60 | 61 | 61 | 68 |
6 months | ||||
Still on treatment in NSR | 38% | 44% | 52% | 32% |
D/C for recurrence | 55% | 49% | 33% | 63% |
D/C for AEs | 3% | 3% | 8% | 4% |
12 months | ||||
Still on treatment in NSR | 32% | 26% | 46% | 22% |
D/C for recurrence | 58% | 57% | 36% | 72% |
D/C for AEs | 7% | 11% | 8% | 6% |
Study 2 | ||||
Randomized | 135 | 133 | 129 | 137 |
Achieved NSR | 103 | 118 | 100 | 106 |
6 months | ||||
Still on treatment in NSR | 41% | 49% | 57% | 22% |
D/C for recurrence | 48% | 42% | 27% | 72% |
D/C for AEs | 9% | 6% | 10% | 4% |
12 months | ||||
Still on treatment in NSR | 25% | 42% | 49% | 16% |
D/C for recurrence | 59% | 47% | 32% | 76% |
D/C for AEs | 11% | 6% | 12% | 5% |
Table 3 and Figures 3 and 4 show, by randomized dose, the effectiveness of TIKOSYN in maintaining NSR using Kaplan Meier analysis, which shows patients remaining on treatment.
| TIKOSYN Dose | Placebo | |||
|---|---|---|---|---|
| 125 mcg BID | 250 mcg BID | 500 mcg BID | ||
| Median time to recurrence of AF/AFl could not be estimated accurately for the 250 mcg BID treatment group in Study 2 and the 500 mcg BID treatment groups in Studies 1 and 2 because TIKOSYN maintained >50% of patients (51%, 58%, and 66%, respectively) in NSR for the 12 months duration of the studies. | ||||
Study 1 | ||||
p-value vs. placebo | P=0.21 | P=0.10 | P<0.001 | |
Median time to recurrence (days) | 31 | 179 | >365 | 27 |
Study 2 | ||||
p-value vs. placebo | P=0.006 | P<0.001 | P<0.001 | |
Median time to recurrence (days) | 182 | >365 | >365 | 34 |
Figure 3: Maintenance of Normal Sinus Rhythm, TIKOSYN Regimen vs. Placebo (Study 1)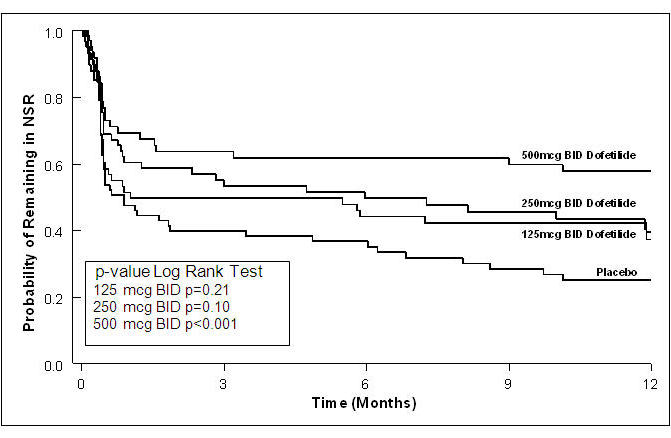
The point estimates of the probabilities of remaining in NSR at 6 and 12 months were 62% and 58%, respectively, for TIKOSYN 500 mcg BID; 50% and 37%, respectively, for TIKOSYN 250 mcg BID; and 37%, and 25%, respectively, for placebo.
Figure 4: Maintenance of Normal Sinus Rhythm, TIKOSYN Regimen vs. Placebo (Study 2)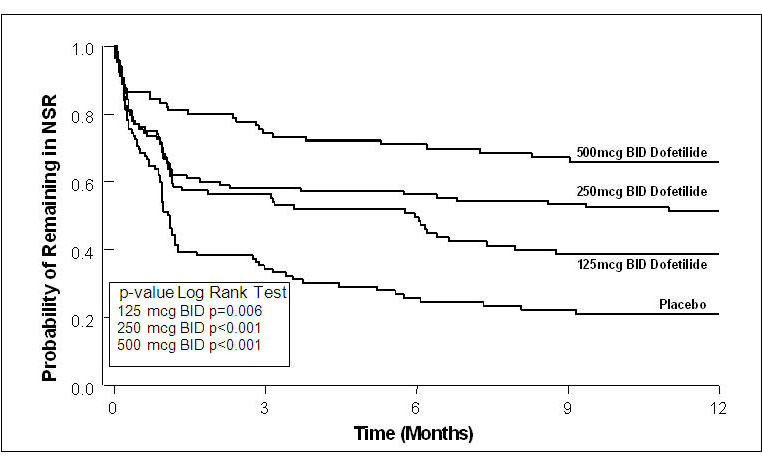
The point estimates of the probabilities of remaining in NSR at 6 and 12 months were 71% and 66%, respectively, for TIKOSYN 500 mcg BID; 56% and 51%, respectively, for TIKOSYN 250 mcg BID; and 26% and 21%, respectively, for placebo.
In both studies, TIKOSYN resulted in a dose-related increase in the number of patients maintained in NSR at all time periods and delayed the time of recurrence of sustained AF. Data pooled from both studies show that there is a positive relationship between the probability of staying in NSR, TIKOSYN dose, and increase in QTc (see Figure 2 in CLINICAL PHARMACOLOGY, Dose-Response and Concentration Response for Increase in QT Interval).
Analysis of pooled data for patients randomized to a TIKOSYN dose of 500 mcg twice daily showed that maintenance of NSR was similar in both males and females, in both patients aged <65 years and patients ≥65 years of age, and in both patients with atrial flutter as a primary diagnosis and those with a primary diagnosis of atrial fibrillation.
During the period of in-hospital initiation of dosing, 23% of patients in Studies 1 and 2 had their dose adjusted downward on the basis of their calculated creatinine clearance, and 3% had their dose down-titrated due to increased QT interval or QTc. Increased QT interval or QTc led to discontinuation of therapy in 3% of patients.
Safety in Patients with Structural Heart Disease: DIAMOND Studies (The Danish Investigations of Arrhythmia and Mortality on Dofetilide)
The two DIAMOND studies were 3-year trials comparing the effects of TIKOSYN and placebo on mortality and morbidity in patients with impaired left ventricular function (ejection fraction ≤35%). Patients were treated for at least one year. One study was in patients with moderate to severe (60% NYHA Class III or IV) congestive heart failure (DIAMOND CHF) and the other was in patients with recent myocardial infarction (DIAMOND MI) (of whom 40% had NYHA Class III or IV heart failure). Both groups were at relatively high risk of sudden death. The DIAMOND trials were intended to determine whether TIKOSYN could reduce that risk. The trials did not demonstrate a reduction in mortality; however, they provide reassurance that, when initiated carefully, in a hospital or equivalent setting, TIKOSYN did not increase mortality in patients with structural heart disease, an important finding because other antiarrhythmics [notably the Class IC antiarrhythmics studied in the Cardiac Arrhythmia Suppression Trial (CAST) and a pure Class III antiarrhythmic, d-sotalol (SWORD)] have increased mortality in post-infarction populations. The DIAMOND trials therefore provide evidence of a method of safe use of TIKOSYN in a population susceptible to ventricular arrhythmias. In addition, the subset of patients with AF in the DIAMOND trials provide further evidence of safety in a population of patients with structural heart disease accompanying the AF. Note, however, that this AF population was given a lower (250 mcg BID) dose (see CLINICAL STUDIES, DIAMOND Patients with Atrial Fibrillation).
In both DIAMOND studies, patients were randomized to 500 mcg BID of TIKOSYN, but this was reduced to 250 mcg BID if calculated creatinine clearance was 40–60 mL/min, if patients had AF, or if QT interval prolongation (>550 msec or >20% increase from baseline) occurred after dosing. Dose reductions for reduced calculated creatinine clearance occurred in 47% and 45% of DIAMOND CHF and MI patients, respectively. Dose reductions for increased QT interval or QTc occurred in 5% and 7% of DIAMOND CHF and MI patients, respectively. Increased QT interval or QTc (>550 msec or >20% increase from baseline) resulted in discontinuation of 1.8% of patients in DIAMOND CHF and 2.5% of patients in DIAMOND MI.
In the DIAMOND studies, all patients were hospitalized for at least 3 days after treatment was initiated and monitored by telemetry. Patients with QTc greater than 460 msec, second or third degree AV block (unless with pacemaker), resting heart rate <50 bpm, or prior history of polymorphic ventricular tachycardia were excluded.
DIAMOND CHF studied 1518 patients hospitalized with severe CHF who had confirmed impaired left ventricular function (ejection fraction ≤35%). Patients received a median duration of therapy of greater than one year. There were 311 deaths from all causes in patients randomized to TIKOSYN (n=762) and 317 deaths in patients randomized to placebo (n=756). The probability of survival at one year was 73% (95% CI: 70% – 76%) in the TIKOSYN group and 72% (95% CI: 69% – 75%) in the placebo group. Similar results were seen for cardiac deaths and arrhythmic deaths. Torsade de Pointes occurred in 25/762 patients (3.3%) receiving TIKOSYN. The majority of cases (76%) occurred within the first 3 days of dosing. In all, 437/762 (57%) of patients on TIKOSYN and 459/756 (61%) on placebo required hospitalization. Of these, 229/762 (30%) of patients on TIKOSYN and 290/756 (38%) on placebo required hospitalization because of worsening heart failure.
DIAMOND MI studied 1510 patients hospitalized with recent myocardial infarction (2–7 days) who had confirmed impaired left ventricular function (ejection fraction ≤35%). Patients received a median duration of therapy of greater than one year. There were 230 deaths in patients randomized to TIKOSYN (n=749) and 243 deaths in patients randomized to placebo (n=761). The probability of survival at one year was 79% (95% CI: 76% – 82%) in the TIKOSYN group and 77% (95% CI: 74% – 80%) in the placebo group. Cardiac and arrhythmic mortality showed a similar result. Torsade de Pointes occurred in 7/749 patients (0.9%) receiving TIKOSYN. Of these, 4 cases occurred within the first 3 days of dosing and 3 cases occurred between Day 4 and the conclusion of the study. In all, 371/749 (50%) of patients on TIKOSYN and 419/761 (55%) on placebo required hospitalization. Of these, 200/749 (27%) of patients on TIKOSYN and 205/761 (27%) on placebo required hospitalization because of worsening heart failure.
DIAMOND Patients with Atrial Fibrillation (the DIAMOND AF subpopulation). There were 506 patients in the two DIAMOND studies who had atrial fibrillation (AF) at entry to the studies (249 randomized to TIKOSYN and 257 randomized to placebo). DIAMOND AF patients randomized to TIKOSYN received 250 mcg BID; 65% of these patients had impaired renal function, so that 250 mcg BID represents the dose they would have received in the AF trials, which would give drug exposure similar to a person with normal renal function given 500 mcg BID. In the DIAMOND AF subpopulation, there were 111 deaths (45%) in the 249 patients in the TIKOSYN group and 116 deaths (45%) in the 257 patients in the placebo group. Hospital readmission rates for any reason were 125/249 or 50% on TIKOSYN and 156/257 or 61% for placebo. Of these, readmission rates for worsening heart failure were 73/249 or 29% on TIKOSYN and 102/257 or 40% for placebo.
Of the 506 patients in the DIAMOND studies who had atrial fibrillation or flutter at baseline, 12% of patients in the TIKOSYN group and 2% of patients in the placebo group had converted to normal sinus rhythm after one month. In those patients converted to normal sinus rhythm, 79% of the TIKOSYN group and 42% of the placebo group remained in normal sinus rhythm for one year.
In the DIAMOND studies, although Torsade de Pointes occurred more frequently in the TIKOSYN-treated patients (see ADVERSE REACTIONS), TIKOSYN, given with an initial 3-day hospitalization and with dose modified for reduced creatinine clearance and increased QT interval, was not associated with an excess risk of mortality in these populations with structural heart disease in the individual studies or in an analysis of the combined studies. The presence of atrial fibrillation did not affect outcome.
Find TIKOSYN® medical information:
Find TIKOSYN® medical information:
TIKOSYN® Quick Finder
Health Professional Information
Clinical Studies
CLINICAL STUDIES
Chronic Atrial Fibrillation and/or Atrial Flutter
Two randomized, parallel, double-blind, placebo-controlled, dose-response trials evaluated the ability of TIKOSYN 1) to convert patients with atrial fibrillation or atrial flutter (AF/AFl) of more than 1 week duration to normal sinus rhythm (NSR) and 2) to maintain NSR (delay time to recurrence of AF/AFl) after drug-induced or electrical cardioversion. A total of 996 patients with a one week to two year history of atrial fibrillation/atrial flutter were enrolled. Both studies randomized patients to placebo or to doses of TIKOSYN 125 mcg, 250 mcg, 500 mcg, or in one study a comparator drug, given twice a day (these doses were lowered based on calculated creatinine clearance and, in one of the studies, for QT interval or QTc). All patients were started on therapy in a hospital where their ECG was monitored (see DOSAGE AND ADMINISTRATION).
Patients were excluded from participation if they had had syncope within the past 6 months, AV block greater than first degree, MI or unstable angina within 1 month, cardiac surgery within 2 months, history of QT interval prolongation or polymorphic ventricular tachycardia associated with use of antiarrhythmic drugs, QT interval or QTc >440 msec, serum creatinine >2.5 mg/mL, significant diseases of other organ systems; used cimetidine; or used drugs known to prolong the QT interval.
Both studies enrolled mostly Caucasians (over 90%), males (over 70%), and patients ≥65 years of age (over 50%). Most (>90%) were NYHA Functional Class I or II. Approximately one-half had structural heart disease (including ischemic heart disease, cardiomyopathies, and valvular disease) and about one-half were hypertensive. A substantial proportion of patients were on concomitant therapy, including digoxin (over 60%), diuretics (over 20%), and ACE inhibitors (over 30%). About 90% were on anticoagulants.
Acute conversion rates are shown in Table 1 for randomized doses (doses were adjusted for calculated creatinine clearance and, in Study 1, for QT interval or QTc). Of patients who converted pharmacologically, approximately 70% converted within 24–36 hours.
| TIKOSYN Dose | Placebo | |||
|---|---|---|---|---|
| 125 mcg BID | 250 mcg BID | 500 mcg BID | ||
Study 1 | 5/82(6%) | 8/82(10%) | 23/77(30%) | 1/84(1%) |
Study 2 | 8/135(6%) | 14/133(11%) | 38/129(29%) | 2/137(1%) |
Patients who did not convert to NSR with randomized therapy within 48–72 hours had electrical cardioversion. Those patients remaining in NSR after conversion in hospital were continued on randomized therapy as outpatients (maintenance period) for up to one year unless they experienced a recurrence of atrial fibrillation/atrial flutter or withdrew for other reasons.
Table 2 shows, by randomized dose, the percentage of patients at 6 and 12 months in both studies who remained on treatment in NSR and the percentage of patients who withdrew because of recurrence of AF/AFl or adverse events.
| TIKOSYN Dose | Placebo | |||
|---|---|---|---|---|
| 125 mcg BID | 250 mcg BID | 500 mcg BID | ||
| Note that columns do not add up to 100% due to discontinuations for "other" reasons. | ||||
Study 1 | ||||
Randomized | 82 | 82 | 77 | 84 |
Achieved NSR | 60 | 61 | 61 | 68 |
6 months | ||||
Still on treatment in NSR | 38% | 44% | 52% | 32% |
D/C for recurrence | 55% | 49% | 33% | 63% |
D/C for AEs | 3% | 3% | 8% | 4% |
12 months | ||||
Still on treatment in NSR | 32% | 26% | 46% | 22% |
D/C for recurrence | 58% | 57% | 36% | 72% |
D/C for AEs | 7% | 11% | 8% | 6% |
Study 2 | ||||
Randomized | 135 | 133 | 129 | 137 |
Achieved NSR | 103 | 118 | 100 | 106 |
6 months | ||||
Still on treatment in NSR | 41% | 49% | 57% | 22% |
D/C for recurrence | 48% | 42% | 27% | 72% |
D/C for AEs | 9% | 6% | 10% | 4% |
12 months | ||||
Still on treatment in NSR | 25% | 42% | 49% | 16% |
D/C for recurrence | 59% | 47% | 32% | 76% |
D/C for AEs | 11% | 6% | 12% | 5% |
Table 3 and Figures 3 and 4 show, by randomized dose, the effectiveness of TIKOSYN in maintaining NSR using Kaplan Meier analysis, which shows patients remaining on treatment.
| TIKOSYN Dose | Placebo | |||
|---|---|---|---|---|
| 125 mcg BID | 250 mcg BID | 500 mcg BID | ||
| Median time to recurrence of AF/AFl could not be estimated accurately for the 250 mcg BID treatment group in Study 2 and the 500 mcg BID treatment groups in Studies 1 and 2 because TIKOSYN maintained >50% of patients (51%, 58%, and 66%, respectively) in NSR for the 12 months duration of the studies. | ||||
Study 1 | ||||
p-value vs. placebo | P=0.21 | P=0.10 | P<0.001 | |
Median time to recurrence (days) | 31 | 179 | >365 | 27 |
Study 2 | ||||
p-value vs. placebo | P=0.006 | P<0.001 | P<0.001 | |
Median time to recurrence (days) | 182 | >365 | >365 | 34 |
Figure 3: Maintenance of Normal Sinus Rhythm, TIKOSYN Regimen vs. Placebo (Study 1)
The point estimates of the probabilities of remaining in NSR at 6 and 12 months were 62% and 58%, respectively, for TIKOSYN 500 mcg BID; 50% and 37%, respectively, for TIKOSYN 250 mcg BID; and 37%, and 25%, respectively, for placebo.
Figure 4: Maintenance of Normal Sinus Rhythm, TIKOSYN Regimen vs. Placebo (Study 2)
The point estimates of the probabilities of remaining in NSR at 6 and 12 months were 71% and 66%, respectively, for TIKOSYN 500 mcg BID; 56% and 51%, respectively, for TIKOSYN 250 mcg BID; and 26% and 21%, respectively, for placebo.
In both studies, TIKOSYN resulted in a dose-related increase in the number of patients maintained in NSR at all time periods and delayed the time of recurrence of sustained AF. Data pooled from both studies show that there is a positive relationship between the probability of staying in NSR, TIKOSYN dose, and increase in QTc (see Figure 2 in CLINICAL PHARMACOLOGY, Dose-Response and Concentration Response for Increase in QT Interval).
Analysis of pooled data for patients randomized to a TIKOSYN dose of 500 mcg twice daily showed that maintenance of NSR was similar in both males and females, in both patients aged <65 years and patients ≥65 years of age, and in both patients with atrial flutter as a primary diagnosis and those with a primary diagnosis of atrial fibrillation.
During the period of in-hospital initiation of dosing, 23% of patients in Studies 1 and 2 had their dose adjusted downward on the basis of their calculated creatinine clearance, and 3% had their dose down-titrated due to increased QT interval or QTc. Increased QT interval or QTc led to discontinuation of therapy in 3% of patients.
Safety in Patients with Structural Heart Disease: DIAMOND Studies (The Danish Investigations of Arrhythmia and Mortality on Dofetilide)
The two DIAMOND studies were 3-year trials comparing the effects of TIKOSYN and placebo on mortality and morbidity in patients with impaired left ventricular function (ejection fraction ≤35%). Patients were treated for at least one year. One study was in patients with moderate to severe (60% NYHA Class III or IV) congestive heart failure (DIAMOND CHF) and the other was in patients with recent myocardial infarction (DIAMOND MI) (of whom 40% had NYHA Class III or IV heart failure). Both groups were at relatively high risk of sudden death. The DIAMOND trials were intended to determine whether TIKOSYN could reduce that risk. The trials did not demonstrate a reduction in mortality; however, they provide reassurance that, when initiated carefully, in a hospital or equivalent setting, TIKOSYN did not increase mortality in patients with structural heart disease, an important finding because other antiarrhythmics [notably the Class IC antiarrhythmics studied in the Cardiac Arrhythmia Suppression Trial (CAST) and a pure Class III antiarrhythmic, d-sotalol (SWORD)] have increased mortality in post-infarction populations. The DIAMOND trials therefore provide evidence of a method of safe use of TIKOSYN in a population susceptible to ventricular arrhythmias. In addition, the subset of patients with AF in the DIAMOND trials provide further evidence of safety in a population of patients with structural heart disease accompanying the AF. Note, however, that this AF population was given a lower (250 mcg BID) dose (see CLINICAL STUDIES, DIAMOND Patients with Atrial Fibrillation).
In both DIAMOND studies, patients were randomized to 500 mcg BID of TIKOSYN, but this was reduced to 250 mcg BID if calculated creatinine clearance was 40–60 mL/min, if patients had AF, or if QT interval prolongation (>550 msec or >20% increase from baseline) occurred after dosing. Dose reductions for reduced calculated creatinine clearance occurred in 47% and 45% of DIAMOND CHF and MI patients, respectively. Dose reductions for increased QT interval or QTc occurred in 5% and 7% of DIAMOND CHF and MI patients, respectively. Increased QT interval or QTc (>550 msec or >20% increase from baseline) resulted in discontinuation of 1.8% of patients in DIAMOND CHF and 2.5% of patients in DIAMOND MI.
In the DIAMOND studies, all patients were hospitalized for at least 3 days after treatment was initiated and monitored by telemetry. Patients with QTc greater than 460 msec, second or third degree AV block (unless with pacemaker), resting heart rate <50 bpm, or prior history of polymorphic ventricular tachycardia were excluded.
DIAMOND CHF studied 1518 patients hospitalized with severe CHF who had confirmed impaired left ventricular function (ejection fraction ≤35%). Patients received a median duration of therapy of greater than one year. There were 311 deaths from all causes in patients randomized to TIKOSYN (n=762) and 317 deaths in patients randomized to placebo (n=756). The probability of survival at one year was 73% (95% CI: 70% – 76%) in the TIKOSYN group and 72% (95% CI: 69% – 75%) in the placebo group. Similar results were seen for cardiac deaths and arrhythmic deaths. Torsade de Pointes occurred in 25/762 patients (3.3%) receiving TIKOSYN. The majority of cases (76%) occurred within the first 3 days of dosing. In all, 437/762 (57%) of patients on TIKOSYN and 459/756 (61%) on placebo required hospitalization. Of these, 229/762 (30%) of patients on TIKOSYN and 290/756 (38%) on placebo required hospitalization because of worsening heart failure.
DIAMOND MI studied 1510 patients hospitalized with recent myocardial infarction (2–7 days) who had confirmed impaired left ventricular function (ejection fraction ≤35%). Patients received a median duration of therapy of greater than one year. There were 230 deaths in patients randomized to TIKOSYN (n=749) and 243 deaths in patients randomized to placebo (n=761). The probability of survival at one year was 79% (95% CI: 76% – 82%) in the TIKOSYN group and 77% (95% CI: 74% – 80%) in the placebo group. Cardiac and arrhythmic mortality showed a similar result. Torsade de Pointes occurred in 7/749 patients (0.9%) receiving TIKOSYN. Of these, 4 cases occurred within the first 3 days of dosing and 3 cases occurred between Day 4 and the conclusion of the study. In all, 371/749 (50%) of patients on TIKOSYN and 419/761 (55%) on placebo required hospitalization. Of these, 200/749 (27%) of patients on TIKOSYN and 205/761 (27%) on placebo required hospitalization because of worsening heart failure.
DIAMOND Patients with Atrial Fibrillation (the DIAMOND AF subpopulation). There were 506 patients in the two DIAMOND studies who had atrial fibrillation (AF) at entry to the studies (249 randomized to TIKOSYN and 257 randomized to placebo). DIAMOND AF patients randomized to TIKOSYN received 250 mcg BID; 65% of these patients had impaired renal function, so that 250 mcg BID represents the dose they would have received in the AF trials, which would give drug exposure similar to a person with normal renal function given 500 mcg BID. In the DIAMOND AF subpopulation, there were 111 deaths (45%) in the 249 patients in the TIKOSYN group and 116 deaths (45%) in the 257 patients in the placebo group. Hospital readmission rates for any reason were 125/249 or 50% on TIKOSYN and 156/257 or 61% for placebo. Of these, readmission rates for worsening heart failure were 73/249 or 29% on TIKOSYN and 102/257 or 40% for placebo.
Of the 506 patients in the DIAMOND studies who had atrial fibrillation or flutter at baseline, 12% of patients in the TIKOSYN group and 2% of patients in the placebo group had converted to normal sinus rhythm after one month. In those patients converted to normal sinus rhythm, 79% of the TIKOSYN group and 42% of the placebo group remained in normal sinus rhythm for one year.
In the DIAMOND studies, although Torsade de Pointes occurred more frequently in the TIKOSYN-treated patients (see ADVERSE REACTIONS), TIKOSYN, given with an initial 3-day hospitalization and with dose modified for reduced creatinine clearance and increased QT interval, was not associated with an excess risk of mortality in these populations with structural heart disease in the individual studies or in an analysis of the combined studies. The presence of atrial fibrillation did not affect outcome.
Health Professional Information
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5Pm ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.