TALZENNA
(talazoparib)
Find TALZENNA medical information:
Find TALZENNA medical information:
TALZENNA Quick Finder
Highlights
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use TALZENNA safely and effectively. See full prescribing information for TALZENNA. TALZENNA (talazoparib) capsules, for oral use Initial U.S. Approval: 2018 RECENT MAJOR CHANGESINDICATIONS AND USAGETALZENNA is a poly (ADP-ribose) polymerase (PARP) inhibitor indicated for: Breast Cancer
HRR Gene-mutated mCRPC
DOSAGE AND ADMINISTRATIONDOSAGE FORMS AND STRENGTHSCapsules: 0.1 mg, 0.25 mg, 0.35 mg, 0.5 mg, 0.75 mg, and 1 mg (3) CONTRAINDICATIONSNone. (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions (≥20%) as a single agent, including laboratory abnormalities, are:
Most common adverse reactions (≥10%) in combination with enzalutamide, including laboratory abnormalities, are:
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSUSE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 2/2024 |
Indications and Usage
1 INDICATIONS AND USAGE
1.1 BRCA-mutated (gBRCAm) HER2-negative Locally Advanced or Metastatic Breast Cancer
TALZENNA is indicated as a single agent for the treatment of adult patients with deleterious or suspected deleterious germline breast cancer susceptibility gene (BRCA)-mutated (gBRCAm) human epidermal growth factor receptor 2 (HER2)-negative locally advanced or metastatic breast cancer. Select patients for therapy based on an FDA-approved companion diagnostic for TALZENNA [see Dosage and Administration (2.1)].
1.2 HRR Gene-mutated mCRPC
TALZENNA is indicated in combination with enzalutamide for the treatment of adult patients with homologous recombination repair (HRR) gene-mutated metastatic castration-resistant prostate cancer (mCRPC) [see Dosage and Administration (2.3)].
Dosage and Administration
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Information on the FDA-approved tests for the detection of genetic mutations is available at http://www.fda.gov/companiondiagnostics.
gBRCAm HER2-negative Locally Advanced or Metastatic Breast Cancer
Select patients for the treatment of advanced breast cancer with TALZENNA based on the presence of germline BRCA mutations [see Indications and Usage (1.1), Clinical Studies (14.1)].
HRR Gene-mutated Metastatic Castration-Resistant Prostate Cancer
Select patients for the treatment of HRR gene-mutated mCRPC with TALZENNA based on the presence of alterations in genes directly or indirectly involved in HRR (ATM, ATR, BRCA1, BRCA2, CDK12, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2, or RAD51C) [see Indications and Usage (1.2), Clinical Studies (14.2)].
An FDA-approved test for the detection of HRR gene mutations for use with TALZENNA is not currently available.
2.2 Recommended Dosage for gBRCAm HER2-negative Locally Advanced or Metastatic Breast Cancer
The recommended dosage of TALZENNA is 1 mg taken orally once daily, until disease progression or unacceptable toxicity.
2.3 Recommended Dosage for HRR Gene-mutated mCRPC
The recommended dosage of TALZENNA is 0.5 mg taken orally once daily in combination with enzalutamide until disease progression or unacceptable toxicity.
Refer to the enzalutamide prescribing information for recommended enzalutamide dosing information.
Patients receiving TALZENNA and enzalutamide should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy.
2.4 Administration
Take TALZENNA with or without food.
Swallow TALZENNA capsules whole. Do not open or dissolve.
If a patient vomits or misses a dose of TALZENNA, instruct them to take the next prescribed dose at the usual time.
2.5 Dosage Modifications for Adverse Reactions
To manage adverse reactions, consider interruption of treatment with or without dose reduction based on severity and clinical presentation. Recommended dose reductions are indicated in Table 1 and Table 2. Treatment with TALZENNA should be discontinued if more than three dose reductions are required.
gBRCAm HER2-negative Locally Advanced or Metastatic Breast Cancer
Dose Reductions | Dose Level |
Recommended starting dose | 1 mg once daily |
First dose reduction | 0.75 mg once daily |
Second dose reduction | 0.5 mg once daily |
Third dose reduction | 0.25 mg once daily |
HRR Gene-mutated mCRPC
Dose Reductions | Dose Level |
Recommended starting dose | 0.5 mg once daily |
First dose reduction | 0.35 mg once daily |
Second dose reduction | 0.25 mg once daily |
Third dose reduction | 0.1 mg once daily |
Refer to the enzalutamide prescribing information for dose modifications for adverse reactions associated with enzalutamide.
gBRCAm HER2-negative Locally Advanced or Metastatic Breast Cancer and HRR Gene-mutated mCRPC
Monitor complete blood counts monthly and as clinically indicated [see Warnings and Precautions (5.2)].
Adverse Reactions | Withhold TALZENNA | Resume TALZENNA |
Hemoglobin <8 g/dL | ≥9 g/dL | Resume TALZENNA at a reduced dose |
Platelet count <50,000/μL | ≥75,000/μL | |
Neutrophil count <1,000/μL | ≥1500/µL | |
Non-hematologic Grade 3 or | ≤Grade 1 | Consider resuming TALZENNA at a reduced dose or discontinue |
2.6 Recommended Dosage in Patients with Renal Impairment
gBRCAm HER2-negative Locally Advanced or Metastatic Breast Cancer
The recommended dosage of TALZENNA for patients with moderate renal impairment (CLcr 30 - 59 mL/min) is 0.75 mg taken orally once daily [see Use in Specific Populations (8.7)].
The recommended dosage of TALZENNA for patients with severe renal impairment (CLcr 15 - 29 mL/min) is 0.5 mg taken orally once daily [see Use in Specific Populations (8.7)].
HRR Gene-mutated mCRPC
The recommended dosage of TALZENNA for patients with moderate renal impairment (CLcr 30 - 59 mL/min) is 0.35 mg taken orally once daily in combination with enzalutamide [see Use in Specific Populations (8.7)].
The recommended dosage of TALZENNA for patients with severe renal impairment (CLcr 15 - 29 mL/min) is 0.25 mg taken orally once daily in combination with enzalutamide [see Use in Specific Populations (8.7)].
2.7 Dosage Modifications for P-glycoprotein Inhibitors
gBRCAm HER2-negative Locally Advanced or Metastatic Breast Cancer
Avoid coadministration of TALZENNA with the following P-glycoprotein (P-gp) inhibitors: itraconazole, amiodarone, carvedilol, clarithromycin, itraconazole, and verapamil. If coadministration of TALZENNA with these P-gp inhibitors cannot be avoided, reduce the dose of TALZENNA to 0.75 mg taken orally once daily. When the P-gp inhibitor is discontinued, increase the dose of TALZENNA (after 3 – 5 half-lives of the P-gp inhibitor) to the dose of TALZENNA that was used before starting the P-gp inhibitor [see Drug Interactions (7.1)].
Monitor for increased adverse reactions and modify the dosage as recommended for adverse reactions when TALZENNA is coadministered with other P-gp inhibitors [see Dosage and Administration (2.5)].
Dosage Forms and Strengths
3 DOSAGE FORMS AND STRENGTHS
Capsule Strength | Description |
0.1 mg | White cap (printed with “Pfizer” in black) and a white body (printed with “TLZ 0.1” in black) |
0.25 mg | Ivory cap (printed with “Pfizer” in black) and a white body (printed with “TLZ 0.25” in black) |
0.35 mg | Ivory cap (printed with “Pfizer” in black) and an ivory body (printed with “TLZ 0.35” in black) |
0.5 mg | Light pink cap (printed with “Pfizer” in black) and a white body (printed with “TLZ 0.5” in black) |
0.75 mg | Light orange cap (printed with “Pfizer” in black) and a white body (printed with “TLZ 0.75” in black) |
1 mg | Light red cap (printed with “Pfizer” in black) and a white body (printed with “TLZ 1” in black) |
Contraindications
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Myelodysplastic Syndrome/Acute Myeloid Leukemia
Myelodysplastic Syndrome/Acute Myeloid Leukemia (MDS/AML), including cases with a fatal outcome, has been reported in patients who received TALZENNA.
Overall, MDS/AML has been reported in 0.4% (3 out of 788) of solid tumor patients treated with TALZENNA as a single agent in clinical studies. In TALAPRO-2, MDS/AML occurred in 2 out of 511 (0.4%) patients treated with TALZENNA and enzalutamide and in 0 out of 517 (0%) patients treated with placebo and enzalutamide [see Adverse Reactions (6.1)]. The durations of TALZENNA treatment in these five patients prior to developing MDS/AML were 0.3, 1, 2, 3, and 5 years, respectively. Most of these patients had received previous chemotherapy with platinum agents and/or other DNA damaging agents including radiotherapy.
Do not start TALZENNA until patients have adequately recovered from hematological toxicity caused by previous chemotherapy. Monitor blood counts monthly during treatment with TALZENNA. For prolonged hematological toxicities, interrupt TALZENNA and monitor blood counts weekly until recovery. If counts do not recover within 4 weeks, refer the patient to a hematologist for further investigations including bone marrow analysis and blood sample for cytogenetics. If MDS/AML is confirmed, discontinue TALZENNA.
5.2 Myelosuppression
Myelosuppression consisting of anemia, neutropenia, and/or thrombocytopenia, have been reported in patients treated with TALZENNA [see Adverse Reactions (6.1)].
Grade ≥3 anemia, neutropenia, and thrombocytopenia were reported, respectively, in 39%, 21%, and 15% of patients receiving TALZENNA as a single agent. Discontinuation due to anemia, neutropenia, and thrombocytopenia occurred, respectively, in 0.7%, 0.3%, and 0.3% of patients.
In TALAPRO-2, Grade ≥3 anemia, neutropenia, and thrombocytopenia were reported, respectively, in 45%, 18%, and 8% of patients receiving TALZENNA and enzalutamide. Overall, 39% of patients (199/511) required a red blood cell transfusion, including 22% (111/511) who required multiple transfusions. Discontinuation due to anemia, neutropenia, and thrombocytopenia occurred, respectively, in 7%, 3%, and 0.4% of patients.
Withhold TALZENNA until patients have adequately recovered from hematological toxicity caused by previous therapy. Monitor blood counts monthly during treatment with TALZENNA. If hematological toxicities do not resolve within 28 days, discontinue TALZENNA and refer the patient to a hematologist for further investigations including bone marrow analysis and blood sample for cytogenetics [see Dosage and Administration (2.5)].
5.3 Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal data, TALZENNA can cause fetal harm when administered to a pregnant woman. In an animal reproduction study, administration of talazoparib to pregnant rats during the period of organogenesis caused fetal malformations and structural skeletal variations, and embryo-fetal death at exposures that were 0.24 times the area under the concentration-time curve (AUC) in patients receiving the recommended human dose of 1 mg daily. Apprise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of TALZENNA [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1)].
Based on findings from genetic toxicity and animal reproduction studies, advise male patients with female partners of reproductive potential or who are pregnant to use effective contraception during treatment and for 4 months following the last dose of TALZENNA [see Use in Specific Populations (8.1, 8.3), Nonclinical Toxicology (13.1)].
Adverse Reactions
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- •
- Myelodysplastic Syndrome/Acute Myeloid Leukemia [see Warnings and Precautions (5.1)]
- •
- Myelosuppression [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in the WARNINGS AND PRECAUTIONS section reflect exposure to single agent TALZENNA in solid tumor clinical studies, including 286 patients enrolled in EMBRACA trial and to TALZENNA 0.5 mg daily in combination with enzalutamide in 511 patients enrolled in the TALAPRO-2 trial that included 197 patients with HRR gene-mutated mCRPC.
gBRCAm HER2-negative Locally Advanced or Metastatic Breast Cancer
EMBRACA
The safety of TALZENNA as a single agent was evaluated in gBRCAm patients with HER2-negative locally advanced or metastatic breast cancer who had previously received no more than 3 lines of chemotherapy for the treatment of locally advanced/metastatic disease [see Clinical Studies (14.1)]. EMBRACA was a randomized, open-label, multi-center study in which 412 patients received either TALZENNA 1 mg once daily (N=286) or a chemotherapy agent (capecitabine, eribulin, gemcitabine, or vinorelbine) of the healthcare provider's choice (N=126) until disease progression or unacceptable toxicity. The median duration of study treatment was 6.1 months in patients who received TALZENNA and 3.9 months in patients who received chemotherapy.
Serious adverse reactions of TALZENNA occurred in 32% of patients. Serious adverse reactions reported in >2% of patients included anemia (6%) and pyrexia (2%). Fatal adverse reactions occurred in 1% of patients, including cerebral hemorrhage, liver disorder, veno-occlusive liver disease, and worsening neurological symptoms (1 patient each).
Permanent discontinuation due to adverse reactions occurred in 5% of TALZENNA patients. Dosing interruptions due to an adverse reaction of any grade occurred in 65% of patients receiving TALZENNA; dose reductions due to any cause occurred in 53% of TALZENNA patients.
The most common (≥20%) adverse reactions, including laboratory abnormalities, were hemoglobin decreased, neutrophils decreased, lymphocytes decreased, platelets decreased, fatigue, glucose increased, aspartate aminotransferase increased, alkaline phosphatase increased, alanine aminotransferase increased, calcium decreased, nausea, headache, vomiting, alopecia, diarrhea, and decreased appetite.
Table 5 and Table 6 summarize the most common adverse reactions and laboratory abnormalities, respectively, in patients treated with TALZENNA or chemotherapy in the EMBRACA study.
| Adverse Reactions | TALZENNA N=286 (%) | Chemotherapy N=126 (%) | ||||
|---|---|---|---|---|---|---|
| Grades 1–4 | Grade 3 | Grade 4 | Grades 1–4 | Grade 3 | Grade 4 | |
| Abbreviation: N=number of patients. | ||||||
General disorders and administration site conditions | ||||||
Fatigue† | 62 | 3 | 0 | 50 | 5 | 0 |
Gastrointestinal disorders | ||||||
Nausea | 49 | 0.3 | 0 | 47 | 2 | 0 |
Vomiting | 25 | 2 | 0 | 23 | 2 | 0 |
Diarrhea | 22 | 1 | 0 | 26 | 6 | 0 |
Nervous system disorders | ||||||
Headache | 33 | 2 | 0 | 22 | 1 | 0 |
Skin and subcutaneous tissue disorders | ||||||
Alopecia | 25 | 0 | 0 | 28 | 0 | 0 |
Metabolism and nutrition disorders | ||||||
Decreased appetite | 21 | 0.3 | 0 | 22 | 1 | 0 |
Clinically relevant adverse reactions in <20% of patients who received TALZENNA included abdominal pain (19%), dizziness (17%), dysgeusia (10%), dyspepsia (10%), stomatitis (8%), and febrile neutropenia (0.3%).
| TALZENNA N*=286 (%) | Chemotherapy N*=126 (%) | |||||
|---|---|---|---|---|---|---|
| Parameter | Grades 1–4 | Grade 3 | Grade 4 | Grades 1–4 | Grade 3 | Grade 4 |
| Abbreviation: N=number of patients. | ||||||
Hemoglobin decreased | 90 | 39 | 0 | 77 | 6 | 0 |
Neutrophils decreased | 68 | 17 | 3 | 70 | 21 | 17 |
Lymphocytes decreased | 76 | 17 | 0.7 | 53 | 8 | 0.8 |
Platelets decreased | 55 | 11 | 4 | 29 | 2 | 0 |
Glucose increased† | 54 | 2 | 0 | 51 | 2 | 0 |
Aspartate aminotransferase Increased | 37 | 2 | 0 | 48 | 3 | 0 |
Alkaline phosphatase increased | 36 | 2 | 0 | 34 | 2 | 0 |
Alanine aminotransferase increased | 33 | 1 | 0 | 37 | 2 | 0 |
Calcium decreased | 28 | 1 | 0 | 16 | 0 | 0 |
HRR Gene-mutated mCRPC
The safety of TALZENNA in combination with enzalutamide was evaluated in patients with HRR gene-mutated mCRPC enrolled in TALAPRO-2 [see Clinical Studies (14.2)]. Patients were randomized to receive either TALZENNA 0.5 mg in combination with enzalutamide 160 mg once daily (n=197), or placebo in enzalutamide 160 mg once daily (n=199) until disease progression or unacceptable toxicity. Among patients receiving TALZENNA, 86% were exposed for 6 months or longer, 60% were exposed for greater than one year, and 18% were exposed for greater than two years.
Serious adverse reactions of TALZENNA in combination with enzalutamide occurred in 30% of patients. Serious adverse reactions reported in >2% of patients included anemia (9%) and fracture (3%). Fatal adverse reactions occurred in 1.5% of patients, including pneumonia, COVID infection, and sepsis (1 patient each).
Permanent discontinuation of TALZENNA due to adverse reactions occurred in 10% of patients treated in the TALZENNA with enzalutamide arm. The most common adverse reactions which resulted in permanent discontinuation of TALZENNA were anemia (4%), fatigue, bone fracture, ischemic heart disease, and spinal cord compression (1% each).
Dosage interruption of TALZENNA due to adverse reactions occurred in 58% of patients treated in the TALZENNA with enzalutamide arm. The most common adverse reactions which resulted in dose interruption of TALZENNA were anemia (42%), neutropenia (15%), and platelet count decreased (9%) and fatigue (5%).
Dose reduction of TALZENNA due to adverse reactions occurred in 52% of patients treated in the TALZENNA with enzalutamide arm. The most common adverse reactions which resulted in dose reduction of TALZENNA were anemia (43%), neutrophil count decreased (15%), platelet count decreased (6%), and fatigue (4%).
The most common adverse reactions (≥10%), including laboratory abnormalities, in patients who received TALZENNA with enzalutamide were hemoglobin decreased, neutrophils decreased, lymphocytes decreased, fatigue, platelets decreased, calcium decreased, nausea, decreased appetite, sodium decreased, phosphate decreased, fractures, magnesium decreased, dizziness, bilirubin increased, potassium decreased, and dysgeusia.
Table 7 and Table 8 summarize the most common adverse reactions and laboratory abnormalities, respectively, in the TALAPRO-2 study.
| Abbreviation: N=number of patients. | ||||||
TALZENNA with Enzalutamide N=197 | Placebo with Enzalutamide N=199 | |||||
Grades 1-4 % | Grade 3 % | Grade 4 % | Grades 1-4 % | Grade 3 % | Grade 4 % | |
Fatigue† | 49 | 4 | 0 | 40 | 1 | 0 |
Nausea | 21 | 2 | 0 | 17 | 1 | 0.5 |
Decreased appetite | 20 | 1 | 0 | 14 | 1 | 1 |
Fractures‡ | 14 | 3 | 0 | 10 | 1.5 | 0 |
Dizziness§ | 13 | 1.5 | 0 | 9 | 1.5 | 0 |
Dysgeusia¶ | 10 | 0 | 0 | 4.5 | 0 | 0 |
Clinically relevant adverse reactions in <10% of patients who received TALZENNA with enzalutamide included abdominal pain (9%), vomiting (9%), alopecia (7%), dyspepsia (4%), venous thromboembolism (3%) and stomatitis (2%).
| Abbreviation: N=number of patients. | ||||||
| ||||||
Laboratory Abnormality | TALZENNA with Enzalutamide N=197* | Placebo with Enzalutamide N=199* | ||||
Grades 1‑4 % | Grade 3 % | Grade 4 % | Grades 1‑4 % | Grade 3 % | Grade 4 % | |
Hemoglobin decreased | 79 | 41 | 0 | 34 | 6 | 0 |
Neutrophils decreased | 60 | 18 | 1 | 18 | 0 | 1 |
Lymphocytes decreased | 58 | 13 | 0 | 36 | 7 | 0 |
Platelets decreased | 45 | 6 | 3 | 8 | 0.5 | 0 |
Calcium decreased | 25 | 0 | 1 | 11 | 0 | 1 |
Sodium decreased | 22 | 3 | 0 | 20 | 1.5 | 0 |
Phosphate decreased | 17 | 3 | 1 | 13 | 2 | 0 |
Magnesium decreased | 14 | 0 | 1 | 12 | 0 | 0.5 |
Bilirubin increased | 11 | 0.5 | 0 | 7 | 0 | 0 |
Potassium decreased | 11 | 0 | 1 | 7 | 1 | 0.5 |
Drug Interactions
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on TALZENNA
Effect of P-gp Inhibitors
Breast Cancer
Avoid coadministration of TALZENNA with the following P-gp inhibitors: itraconazole, amiodarone, carvedilol, clarithromycin, itraconazole, and verapamil. If coadministration of TALZENNA with these P-gp inhibitors cannot be avoided, reduce the dose of TALZENNA [see Dosage and Administration (2.7)]. When the P-gp inhibitor is discontinued, increase the dose of TALZENNA [see Dosage and Administration (2.7)].
Coadministration of TALZENNA with these P-gp inhibitors increased talazoparib concentrations [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions.
Monitor for increased adverse reactions and modify the dosage as recommended for adverse reactions when TALZENNA is coadministered with other P-gp inhibitors [see Dosage and Administration (2.5)].
HRR Gene-mutated mCRPC
The effect of coadministration of P-gp inhibitors on talazoparib exposure when TALZENNA is taken in combination with enzalutamide has not been studied. Monitor patients for increased adverse reactions and modify the dosage as recommended for adverse reactions when TALZENNA is coadministered with a P-gp inhibitor [see Dosage and Administration (2.5)].
Effect of BCRP Inhibitors
Monitor patients for increased adverse reactions and modify the dosage as recommended for adverse reactions when TALZENNA is coadministered with a BCRP inhibitor [see Dosage and Administration (2.5)].
Coadministration of TALZENNA with BCRP inhibitors may increase talazoparib exposure [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions.
Use in Specific Populations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], TALZENNA can cause embryo-fetal harm when administered to a pregnant woman. There are no available data on TALZENNA use in pregnant women to inform a drug-associated risk. In an animal reproduction study, the administration of talazoparib to pregnant rats during the period of organogenesis caused fetal malformations and structural skeletal variations and embryo-fetal death at maternal exposures that were 0.24 times the AUC in patients receiving the recommended dose of 1 mg daily (see Data). Apprise pregnant women and females of reproductive potential of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the general U.S. population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2% to 4% and 15% to 20%, respectively.
Animal Data
In an embryo-fetal development toxicity study, pregnant rats received oral doses of 0.015, 0.05, and 0.15 mg/kg/day talazoparib during the period of organogenesis. Talazoparib caused embryo-fetal death at doses ≥0.015 mg/kg/day (approximately 0.24 times the AUC in patients at the recommended dose of 1 mg daily). A dose of 0.015 mg/kg/day caused decreased fetal body weights and an increased incidence of fetal malformations (depressed eye bulge, small eye, split sternebra, and fused cervical vertebral arch) and structural variations including misshapen or incomplete ossification of the sternebra, skull, rib, and vertebra.
8.2 Lactation
Risk Summary
There are no data on the presence of talazoparib in human milk, the effects of the drug on milk production, or the effects of the drug on the breastfed child. Because of the potential for serious adverse reactions in a breastfed child from talazoparib, advise lactating women not to breastfeed during treatment with TALZENNA and for 1 month after the final dose.
8.3 Females and Males of Reproductive Potential
TALZENNA can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating TALZENNA treatment.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment and for 7 months following the last dose of TALZENNA.
Males
Based on genotoxicity and animal reproduction studies, advise male patients with female partners of reproductive potential and pregnant partners to use effective contraception during treatment with TALZENNA and for 4 months following the last dose [see Use in Specific Populations (8.1), Nonclinical Toxicology (13.1)].
Infertility
Males
Based on animal studies, TALZENNA may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of TALZENNA have not been established in pediatric patients.
8.5 Geriatric Use
In clinical trials of TALZENNA enrolling 494 patients with advanced solid tumors who received TALZENNA 1 mg daily as a single agent, 85 (17%) patients were ≥65 years of age, and this included 19 (4%) patients who were ≥75 years old. There were 5 patients ≥85 years old. In the TALAPRO-2 trial, of 197 patients who received TALZENNA, 77% were ≥65 years of age, while 30% were ≥75 years of age. No overall differences in safety or effectiveness of TALZENNA were observed between these patients and younger patients.
8.6 Hepatic Impairment
No dosage modification is recommended for patients with hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
Reduce the recommended dosage of TALZENNA in patients with moderate (CLcr 30 – 59 mL/min) and severe (CLcr 15 – 29 mL/min) renal impairment [see Dosage and Administration (2.7)]. Monitor patients with severe renal impairment for increased adverse reactions and modify the dosage as recommended for adverse reactions [see Dosage and Administration (2.5)].
No dose adjustment is recommended for patients with mild renal impairment (CLcr 60 – 89 mL/min). TALZENNA has not been studied in patients requiring hemodialysis.
Overdosage
Description
11 DESCRIPTION
Talazoparib is an inhibitor of mammalian polyadenosine 5'-diphosphoribose (ADP-ribose) polymerase (PARP) enzymes. The chemical name of talazoparib tosylate is (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyrido[4,3,2-de]phthalazin-3-one 4-methylbenzenesulfonate (1:1). The chemical formula of talazoparib tosylate is C26H22F2N6O4S, and the relative molecular mass is 552.56 Daltons. The chemical structure of talazoparib tosylate is shown below:
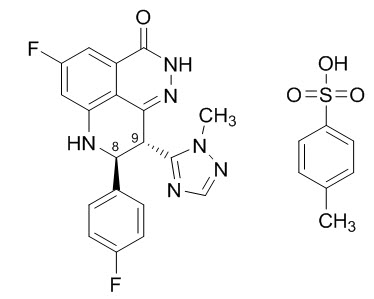
Talazoparib tosylate is a white to yellow solid. TALZENNA capsules for oral use are available as:
- •
- 0.1 mg hard hypromellose (HPMC) capsule that contains 0.145 mg talazoparib tosylate equivalent to 0.1 mg talazoparib free base, or
- •
- 0.25 mg HPMC capsule that contains 0.363 mg talazoparib tosylate equivalent to 0.25 mg talazoparib free base, or
- •
- 0.35 mg HPMC capsule that contains 0.509 mg talazoparib tosylate equivalent to 0.35 mg talazoparib free base, or
- •
- 0.5 mg HPMC capsule that contains 0.727 mg talazoparib tosylate equivalent to 0.5 mg talazoparib free base, or
- •
- 0.75 mg HPMC capsule that contains 1.09 mg talazoparib tosylate equivalent to 0.75 mg talazoparib free base, or
- •
- 1 mg HPMC capsule that contains 1.453 mg talazoparib tosylate equivalent to 1 mg talazoparib free base.
Inactive ingredients: silicified microcrystalline cellulose (sMCC). The capsule shells contain hypromellose (HPMC), yellow iron oxide, red iron oxide and titanium dioxide; and the printing ink contains shellac, black iron oxide, potassium hydroxide, ammonium hydroxide, and propylene glycol.
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Talazoparib is an inhibitor of PARP enzymes, including PARP1 and PARP2, which play a role in DNA repair. In vitro studies with cancer cell lines that harbored defects in DNA repair genes, including BRCA1 and BRCA2, have shown that talazoparib-induced cytotoxicity may involve inhibition of PARP enzymatic activity and increased formation of PARP-DNA complexes resulting in DNA damage, decreased cell proliferation, and apoptosis. Talazoparib anti-tumor activity was observed in patient-derived xenograft breast cancer models bearing mutated BRCA1 or mutated BRCA2 or wild type BRCA1 and BRCA2.
12.2 Pharmacodynamics
The exposure-response relationship and time course of pharmacodynamic response for the safety and effectiveness of TALZENNA have not been fully characterized.
12.3 Pharmacokinetics
After administration of TALZENNA 1 mg orally once daily as a single agent (the recommended dosage for breast cancer), the mean [% coefficient of variation (CV%)] AUC and maximum observed plasma concentration (Cmax) of talazoparib at steady-state was 208 (37%) ng.hr/mL and 16.4 (32%) ng/mL, respectively. The mean (CV%) steady-state Ctrough was 3.53 (61%) ng/mL.
After administration of TALZENNA 0.5 mg orally once daily (the recommended dosage for prostate cancer) in combination with enzalutamide, the mean (CV%) steady-state Ctrough ranged from 3.29 to 3.68 ng/mL (45% to 48%).
The pharmacokinetics (PK) of talazoparib is linear from 0.025 mg to 2 mg (2 times the recommended dose for breast cancer). The median accumulation ratio of talazoparib following 1 mg orally once daily is 2.3 to 5.2. Talazoparib plasma concentrations reached steady-state within 2 to 3 weeks when administered as a single agent and within 9 weeks when coadministered with enzalutamide.
Absorption
The median time to Cmax (Tmax) was generally between 1 to 2 hours after dosing.
Food Effect
Following a single TALZENNA 0.5 mg dose with high-fat, high-calorie food (approximately 800 to 1000 calories with 150, 250, and 500 to 600 calories from protein, carbohydrate, and fat, respectively), the mean Cmax was decreased by 46%, the median Tmax was delayed from 1 to 4 hours, and AUC was not affected.
Distribution
The mean apparent volume of distribution of talazoparib is 420 L. In vitro, protein binding of talazoparib is 74% and is independent of talazoparib concentration.
Elimination
The mean terminal plasma half-life (±standard deviation) is 90 (±58) hours and the mean apparent oral clearance (inter-subject variability) is 6.45 L/h (31%).
Specific Populations
Age (18 to 88 years), sex, race (361 White, 41 Asian, 16 Black, 9 Others, and 63 Not Reported), body weight (36 to 162 kg), and mild to severe hepatic impairment had no clinically significant effect on the PK of talazoparib.
Patients with Renal Impairment
Mild (eGFR 60 – 89 mL/min/1.73 m2) renal impairment had no clinically significant effect on talazoparib pharmacokinetics. Talazoparib steady-state total exposure (AUC) increased by 43% in subjects with moderate (eGFR 30 – 59 mL/min/1.73 m2) renal impairment and 163% in patients with severe (eGFR 15 – 29 mL/min/1.73 m2) renal impairment relative to subjects with normal renal function (eGFR ≥ 90 mL/min/1.73 m2). Talazoparib steady-state peak concentration (Cmax) increased by 32% in subjects with moderate renal impairment and 89% in subjects with severe renal impairment, relative to subjects with normal renal function. Similar increases in AUC were observed with talazoparib when given in combination with enzalutamide for patients with moderate and severe renal impairment. The PK of talazoparib has not been studied in patients requiring hemodialysis. There was no evidence of a relationship between the protein binding of talazoparib and renal function.
Drug Interaction Studies
Clinical Studies
Effect of P-gp Inhibitors: Coadministration of a P-gp inhibitor (itraconazole) with a single 0.5 mg dose of TALZENNA increased talazoparib AUC and Cmax by approximately 56% and 40%, respectively. Coadministration with the following other P-gp inhibitors: amiodarone, carvedilol, clarithromycin, itraconazole, and verapamil increased talazoparib exposure by 45%.
Coadministration with other P-gp inhibitors (including azithromycin, atorvastatin, diltiazem, felodipine, fluvoxamine, and quercetin) had no clinically significant effect on talazoparib pharmacokinetics.
Effect of P-gp Inducers: Coadministration of a P-gp inducer (rifampin) with a single 1 mg dose of TALZENNA increased talazoparib Cmax by 37% with no effect on talazoparib AUC.
Effect of Acid-Reducing Agents: Coadministration of acid-reducing agents including proton pump inhibitors (PPI), histamine receptor 2 antagonists (H2RA), or other acid reducing agents has no effect on the absorption of talazoparib.
Enzalutamide: Coadministration of enzalutamide with TALZENNA increased talazoparib exposure approximately 2-fold.
In Vitro Studies
Transporters: Talazoparib is a substrate of P-gp and BCRP transporters, but not a substrate of OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, BSEP, MATE1, or MATE2-K.
Talazoparib is not an inhibitor of P-gp, BCRP, OATP1B1, OATP1B3, OCT1, OCT2, OAT1, OAT3, BSEP, MATE1, or MATE2-K.
CYP Enzymes: Talazoparib is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4/5.
Talazoparib is not an inducer of CYP1A2, CYP2B6, or CYP3A4.
UGT: Talazoparib is not an inhibitor of UGT isoforms (1A1, 1A4, 1A6, 1A9, 2B7, and 2B15).
Nonclinical Toxicology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with talazoparib.
Talazoparib was clastogenic in an in vitro chromosomal aberration assay in human peripheral blood lymphocytes and in an in vivo bone marrow micronucleus assay in rats. This clastogenicity is consistent with genomic instability resulting from the primary pharmacology of talazoparib, indicating the potential for genotoxicity in humans. Talazoparib was not mutagenic in a bacterial reverse mutation (Ames) test.
Fertility studies in animals have not been conducted with talazoparib. In repeat-dose toxicity studies up to 3-months duration, talazoparib-related findings in the testis and epididymis at doses ≥0.04 mg/kg/day in rats and ≥0.01 mg/kg/day in dogs included decreased organ weights, luminal cellular debris, reduced sperm, and degeneration/atrophy. These doses in rats and dogs resulted in approximately 1.0 times and 0.2 times, respectively, the exposure (AUC) in humans at the recommended dose of 1 mg daily. Follicular atresia of the ovary was observed in rats at doses ≥1 mg/kg/day talazoparib, approximately 9.5 times the AUC in patients at the recommended dose of 1 mg daily.
Clinical Studies
14 CLINICAL STUDIES
14.1 Deleterious or Suspected Deleterious Germline BRCA-mutated HER2-negative Locally Advanced or Metastatic Breast Cancer
EMBRACA (NCT01945775) was an open label study in which patients (N=431) with gBRCAm HER2-negative locally advanced or metastatic breast cancer were randomized 2:1 to receive TALZENNA 1 mg or healthcare provider's choice of chemotherapy (capecitabine, eribulin, gemcitabine, or vinorelbine) until disease progression or unacceptable toxicity. Randomization was stratified by prior lines of chemotherapy for metastatic disease (0 versus 1, 2, or 3), by triple-negative disease status [triple-negative breast cancer (TNBC) versus non-TNBC], and history of central nervous system (CNS) metastasis (yes versus no).
Patients received no more than 3 prior cytotoxic chemotherapy regimens for their metastatic or locally advanced disease. Patients were required to have received treatment with an anthracycline and/or a taxane (unless contraindicated) in the neoadjuvant, adjuvant, and/or metastatic treatment setting. First-line treatment for advanced or metastatic disease with no prior adjuvant chemotherapy was allowed if the investigator determined that 1 of the 4 chemotherapy choices in the control arm would be an appropriate treatment option for the patient. Patients with prior platinum therapy for advanced disease were required to have no evidence of disease progression during platinum therapy. No prior treatment with a PARP inhibitor was permitted. Of the 431 patients randomized in the EMBRACA study, 408 (95%) were centrally confirmed to have a deleterious or suspected deleterious gBRCAm using a clinical trial assay; out of which 354 (82%) were confirmed using the BRACAnalysis CDx. BRCA mutation status [breast cancer susceptibility gene 1 (BRCA1)-positive or breast cancer susceptibility gene 2 (BRCA2)-positive] was similar across both treatment arms.
The median age of patients treated with TALZENNA was 46 years (range 28 to 84) and 51 years (range 24 to 89) among patients treated with chemotherapy. Among all randomized patients, 1% versus 2% were males, 67% versus 75% were White; 11% versus 11% were Asian, and 4% versus 1% were Black or African American in the TALZENNA and chemotherapy arms, respectively. Almost all patients (98%) in both arms had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Approximately 56% of patients had estrogen receptor-positive and/or progesterone receptor-positive disease; 44% of patients had triple-negative disease, and the proportions were balanced across both treatment arms. Fifteen percent (15%) of patients in the TALZENNA arm and 14% of patients in the chemotherapy arm had a history of CNS metastases. Ninety-one percent (91%) of patients in the TALZENNA arm had received prior taxane therapy, and 85% had received prior anthracycline therapy in any setting. Sixteen percent (16%) of patients in the TALZENNA arm and 21% of patients in the chemotherapy arm had received prior platinum treatment in any setting. The median number of prior cytotoxic regimens for patients with advanced breast cancer was one; 38% received no prior cytotoxic regimens for advanced or metastatic disease, 37% received one, 20% received two, and 5% received three or more prior cytotoxic regimens.
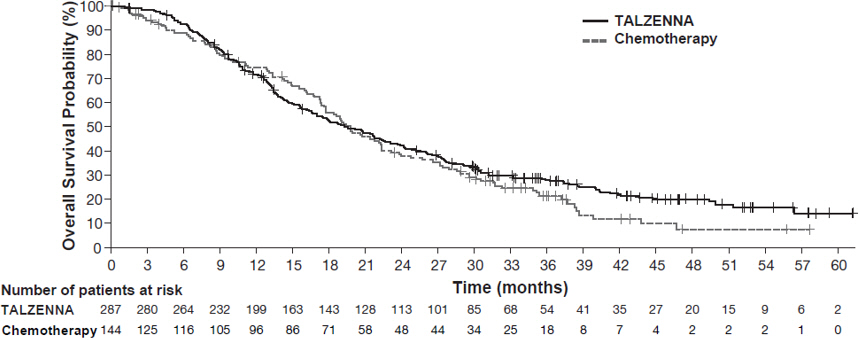
The major efficacy outcome measure was progression-free survival (PFS) evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, as assessed by blinded independent central review (BICR). A statistically significant improvement in PFS was demonstrated for TALZENNA compared with chemotherapy. A sensitivity analysis of investigator-assessed PFS was consistent with the BICR-assessed PFS results. Consistent PFS results were observed across patient subgroups defined by study stratification factors (prior lines of chemotherapy, TNBC status, and history of CNS metastases). Efficacy data from the EMBRACA study are summarized in Table 9, and the Kaplan-Meier curves for PFS are shown in Figure 1 and final overall survival (OS) in Figure 2.
| Abbreviations: BICR=blinded independent central review; CI=confidence interval; DOR=duration of response; ITT=intent-to-treat; N=number of patients; ORR=objective response rate; OS=overall survival; PFS=progression-free survival. | ||
| ||
TALZENNA | Chemotherapy | |
PFS by BICR | N=287 | N=144 |
Disease progression or deaths, n (%) | 186 (65) | 83 (58) |
Median months (95% CI) | 8.6 (7.2, 9.3) | 5.6 (4.2, 6.7) |
Hazard ratio (95% CI)* | 0.54 (0.41, 0.71) | |
p-value† | p<0.0001 | |
Patients with measurable disease by investigator‡ | N=219 | N=114 |
ORR, % (95% CI)§ | 50.2 (43.4, 57.0) | 18.4 (11.8, 26.8) |
Median¶ DOR months (95% CI) | 6.4 (5.4, 9.5) | 3.9 (3.0, 7.6) |
OS | N=287 | N=144 |
Deaths, n (%) | 216 (75) | 108 (75) |
Median months (95% CI) | 19.3 (16.6, 22.5) | 19.5 (17.4, 22.4) |
Hazard ratio (95% CI)* | 0.85 (0.67, 1.07) | |
p-value† | p=0.1693 | |
Figure 1. Kaplan-Meier Curves of PFS – EMBRACA Study
| Abbreviation: PFS=progression-free survival. |
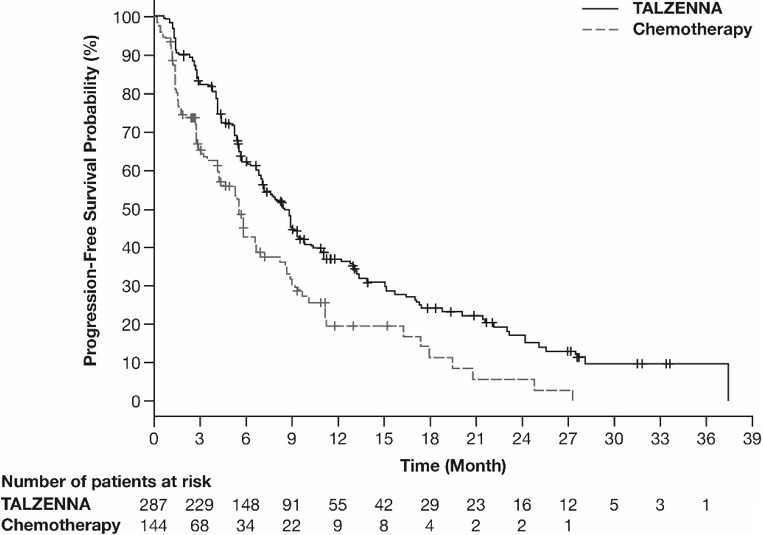 |
Figure 2. Kaplan-Meier Curves of OS – EMBRACA Study (ITT Population)
| Abbreviations: ITT=intent-to-treat; OS=overall survival. |
 |
14.2 HRR Gene-mutated mCRPC
The efficacy of TALZENNA in combination with enzalutamide was evaluated in TALAPRO-2 (NCT03395197), a randomized, double-blind, placebo-controlled, multi-cohort trial in which 399 patients with HRR gene-mutated (HRRm) mCRPC were randomized 1:1 to receive enzalutamide 160 mg daily plus either TALZENNA 0.5 mg or placebo daily until unacceptable toxicity or progression. All patients received a GnRH analog or had prior bilateral orchiectomy and needed to have progressed on prior androgen deprivation therapy. Prior treatment with a CYP17 inhibitor or docetaxel for metastatic castration-sensitive prostate cancer (mCSPC) was permitted. Mutation status of HRR genes was determined prospectively using solid tumor tissue or circulating tumor DNA (ctDNA)-based next generation sequencing assays. Patients were required to have a mutation in at least one of 12 genes involved directly or indirectly in the HRR pathway (ATM, ATR, BRCA1, BRCA2, CDK12, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2, or RAD51C).
Randomization was stratified by previous treatment with a CYP17 inhibitor or docetaxel (yes/no).
The median age was 70 years (range: 41 to 90); 100% were male; 68% were White, 21% Asian, 2.8% Black, 0.8% Other, 7% unknown/not reported; 12% were Hispanic/Latino; and baseline ECOG performance status was 0 (62%) or 1 (38%). Thirty-nine percent of patients had bone-only disease; 15% had visceral disease. In the mCSPC setting, 29% percent of patients had received docetaxel and 9% had received a prior CYP17 inhibitor. The most commonly mutated HRR genes (>5%), including co-occurring mutations, were: BRCA2 (34%), ATM (22%), CDK12 (19%), CHEK2 (18%), and BRCA1 (6%).
The major efficacy outcome measure was radiographic progression-free survival (rPFS) evaluated according to RECIST, version 1.1 and Prostate Cancer Working Group (PCWG3) (bone) criteria, assessed by BICR. An additional efficacy outcome measure was OS.
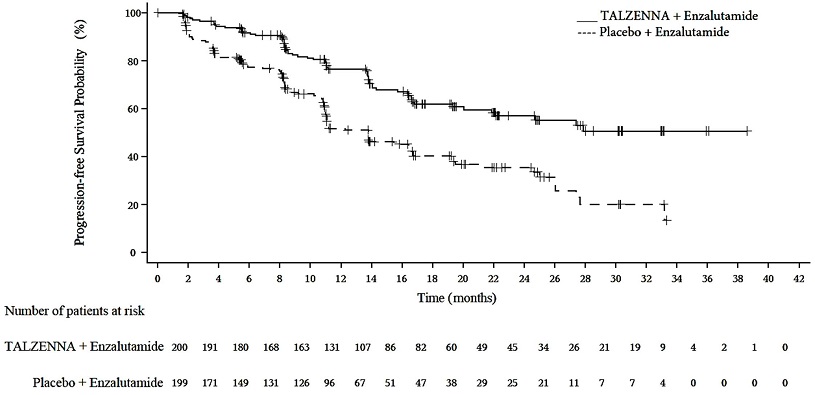
A statistically significant improvement in rPFS was demonstrated at the pre-specified interim analysis in patients randomized to TALZENNA in combination with enzalutamide compared with placebo in combination with enzalutamide. Consistent rPFS results were observed in patients who received or did not receive a prior CYP17 inhibitor or docetaxel. The OS data were not mature at the time of the rPFS analysis (24% of patients had died). Efficacy results are presented in Table 10 and Figure 3.
| Abbreviations: BICR=blinded independent central review; CI=confidence interval; CSPC=castration-sensitive prostate cancer; HRRm=homologous recombination repair gene-mutated; mCRPC=metastatic castration-resistant prostate cancer; N=number of patients; NE=not evaluable. | ||
TALZENNA with Enzalutamide (N=200) | Placebo with Enzalutamide (N=199) | |
Radiographic Progression-free Survival (rPFS) by BICR | ||
Number of rPFS events, n (%) | 66 (33) | 104 (52) |
Median months (95% CI) | NE (21.9, NE) | 13.8 (11.0, 16.7) |
Hazard ratio (95% CI)* | 0.45 (0.33, 0.61) | |
p-value† | <0.0001 | |
Figure 3. Kaplan-Meier Curve for rPFS in TALAPRO-2 (HRR Gene-mutated mCRPC)
| Abbreviations: HRRm=homologous recombination repair gene-mutated; mCRPC=metastatic castration-resistant prostate cancer; rPFS=radiographic progression-free survival. |
 |
Exploratory subgroup analyses of rPFS for patients with BRCA-mutated (BRCAm) and non-BRCAm HRRm are presented in Table 11.
| Abbreviations: BRCAm=breast cancer susceptibility gene-mutated; CI=confidence interval; HRRm=homologous recombination repair gene-mutated; NE=not evaluable; rPFS=radiographic progression-free survival. | ||||
| ||||
BRCAm | Non-BRCAm HRRm* | |||
TALZENNA with Enzalutamide N=71 | Placebo with Enzalutamide N=84 | TALZENNA with Enzalutamide N=129 | Placebo with Enzalutamide N=115 | |
rPFS | ||||
Number of events, n (%) | 15 (21) | 54 (64) | 51 (40) | 50 (43) |
Median months (95% CI) | NE (NE, NE) | 11.0 (8.3, 11.1) | 24.7 (16.4, NE) | 16.7 (13.8, 27.7) |
Hazard ratio (95% CI) | 0.20 (0.11, 0.36) | 0.72 (0.49, 1.07) | ||
How Supplied/Storage and Handling
16 HOW SUPPLIED/STORAGE AND HANDLING
TALZENNA is supplied in strengths and package configurations as described in Table 12:
Package Configuration | Capsule Strength (mg) | NDC | |
Bottles of 30 capsules | 0.1 | NDC 0069-1031-30 | White cap (printed with “Pfizer” in black) and a white body (printed with “TLZ 0.1” in black). |
Bottles of 30 capsules | 0.25 | NDC 0069-0296-30 | Ivory cap (printed with "Pfizer" in black) and a white body (printed with "TLZ 0.25" in black). |
Bottles of 30 capsules | 0.35 | NDC 0069-1235-30 | Ivory cap (printed with “Pfizer” in black) and an ivory body (printed with “TLZ 0.35” in black). |
Bottles of 30 capsules | 0.5 | NDC 0069-1501-30 | Light pink cap (printed with "Pfizer" in black) and a white body (printed with "TLZ 0.5" in black). |
Bottles of 30 capsules | 0.75 | NDC 0069-1751-30 | Light orange cap (printed with "Pfizer" in black) and a white body (printed with "TLZ 0.75" in black). |
Bottles of 30 capsules | 1 | NDC 0069-1195-30 | Light red cap (printed with "Pfizer" in black) and a white body (printed with "TLZ 1" in black). |
Medication Guide
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
- •
- MDS/AML: Advise patients to contact their healthcare provider if they experience weakness, feeling tired, fever, weight loss, frequent infections, bruising, bleeding easily, breathlessness, blood in urine or stool, and/or laboratory findings of low blood cell counts, or a need for blood transfusions. This may be a sign of hematological toxicity or a more serious uncommon bone marrow problem called MDS or AML, which have been reported in patients who received PARP inhibitors [see Warnings and Precautions (5.1)].
- •
- Myelosuppression: Advise patients that TALZENNA may affect hematopoiesis and can cause anemia, leukopenia/neutropenia, and/or thrombocytopenia [see Warnings and Precautions (5.2)].
- •
- Administration Instructions: Advise patients that TALZENNA can be taken once daily with or without food. Instruct patients that if they miss a dose of TALZENNA, they should take their next normal dose at the usual time. Also advise patients to swallow each capsule whole, and that capsules must not be opened or dissolved [see Dosage and Administration (2.4)].
- •
- Embryo-Fetal Toxicity: Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with TALZENNA and for 7 months after the last dose. Advise male patients with female partners of reproductive potential or who are pregnant to use effective contraception during treatment and for 4 months after receiving the last dose of TALZENNA [see Warnings and Precautions (5.3), Use in Specific Populations (8.1, 8.3)].
- •
- Lactation: Advise patients not to breastfeed while taking TALZENNA and for 1 month after receiving the last dose [see Use in Specific Populations (8.2)].
| This Patient Information has been approved by the U.S. Food and Drug Administration Revised: 2/2024 | |
PATIENT INFORMATION | |
What is the most important information I should know about TALZENNA? Bone marrow problems called Myelodysplastic Syndrome (MDS) or Acute Myeloid Leukemia (AML). Some people who have cancer and who have received previous treatment with chemotherapy or certain other medicines for their cancer have developed MDS or AML during or after treatment with TALZENNA. MDS or AML may lead to death. If you develop MDS or AML, your healthcare provider will stop treatment with TALZENNA. | |
|
|
Your healthcare provider will do blood tests to check your blood cell counts:
See "What are the possible side effects of TALZENNA?" below for other side effects of TALZENNA. | |
What is TALZENNA?
Your healthcare provider will perform a test to make sure that TALZENNA is right for you. It is not known if TALZENNA is safe and effective in children. | |
Before taking TALZENNA, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription medicines, over-the-counter medicines, vitamins, and herbal supplements. Taking TALZENNA and certain other medicines can affect how TALZENNA works and may cause side effects. | |
How should I take TALZENNA?
| |
What are the possible side effects of TALZENNA? TALZENNA may cause serious side effects, including: The most common side effects of TALZENNA when taken alone include: | |
|
|
The most common side effects of TALZENNA when taken in combination with enzalutamide include: | |
|
|
TALZENNA may cause fertility problems in males. This may affect your ability to father a child. Talk to your healthcare provider if this is a concern for you. These are not all of the possible side effects of TALZENNA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |
How should I store TALZENNA?
Keep TALZENNA and all medicines out of the reach of children. | |
General information about the safe and effective use of TALZENNA. | |
What are the ingredients in TALZENNA? 
| |
Other
This product's labeling may have been updated. For the most recent prescribing information, please visit http://www.pfizer.com.

LAB-1271-8.0
What is the most important information I should know about TALZENNA?
What is the most important information I should know about TALZENNA?
TALZENNA may cause serious side effects, including:Bone marrow problems called Myelodysplastic Syndrome (MDS) or Acute Myeloid Leukemia (AML). Some people who have cancer and who have received previous treatment with chemotherapy or certain other medicines for their cancer have developed MDS or AML during or after treatment with TALZENNA. MDS or AML may lead to death. If you develop MDS or AML, your healthcare provider will stop treatment with TALZENNA.
Symptoms of low blood cell counts are common during treatment with TALZENNA, but can be a sign of serious problems, including MDS or AML. Tell your healthcare provider if you have any of the following symptoms during treatment with TALZENNA:
- •
- weakness
- •
- weight loss
- •
- fever
- •
- frequent infections
- •
- blood in urine or stool
- •
- shortness of breath
- •
- feeling very tired
- •
- bruising or bleeding more easily
Your healthcare provider will do blood tests to check your blood cell counts:
- •
- every month during treatment with TALZENNA.
- •
- weekly if you have low blood cell counts that last a long time. Your healthcare provider may stop treatment with TALZENNA until your blood cell counts improve.
See "What are the possible side effects of TALZENNA?" below for other side effects of TALZENNA.
What is TALZENNA?
What is TALZENNA?
- •
- alone to treat adults with a certain type of breast cancer (human epidermal growth factor receptor 2 [HER2]-negative)
- o
- who have an abnormal inherited BRCA gene, and
- o
- whose cancer has spread to other parts of the body (locally advanced or metastatic).
- •
- in combination with a medicine called enzalutamide, to treat adults with prostate cancer
- o
- with certain abnormal inherited or acquired genes called homologous recombination repair (HRR genes) and
- o
- which no longer responds to a hormone therapy or surgical treatment to lower testosterone and has spread to other parts of the body (metastatic).
Your healthcare provider will perform a test to make sure that TALZENNA is right for you.
It is not known if TALZENNA is safe and effective in children.
Before taking TALZENNA, tell your healthcare provider about all of your medical conditions, including if you:
- •
- have kidney problems.
- •
- are pregnant or plan to become pregnant. TALZENNA can harm your unborn baby, and may cause loss of pregnancy (miscarriage). You should not become pregnant during treatment with TALZENNA. Tell your healthcare provider right away if you are pregnant or become pregnant during treatment with TALZENNA.
- o
- If you are able to become pregnant, your healthcare provider may do a pregnancy test before you start treatment with TALZENNA.
- o
- Females who are able to become pregnant should use effective birth control (contraception) during treatment with TALZENNA and for at least 7 months after receiving the last dose of TALZENNA. Talk to your healthcare provider about forms of birth control that may be right for you.
- o
- Males with female partners who are pregnant or are able to become pregnant should use effective birth control during treatment with TALZENNA and for at least 4 months after receiving the last dose of TALZENNA.
- •
- are breastfeeding or plan to breastfeed. It is not known if TALZENNA passes into your breast milk. Do not breastfeed during treatment with TALZENNA and for at least 1 month after receiving the last dose of TALZENNA. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take, including prescription medicines, over-the-counter medicines, vitamins, and herbal supplements. Taking TALZENNA and certain other medicines can affect how TALZENNA works and may cause side effects.
Know the medicines you take. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take TALZENNA?
How should I take TALZENNA?
- •
- Take TALZENNA exactly as your healthcare provider tells you.
- •
- Do not change your dose or stop taking TALZENNA without first talking with your healthcare provider.
- •
- For breast cancer, take TALZENNA 1 time a day.
- •
- For prostate cancer, take TALZENNA in combination with enzalutamide 1 time a day.
- •
- You should start or continue a gonadotropin-releasing hormone (GnRH) analog therapy during your treatment with TALZENNA and enzalutamide unless you have had a surgery to lower the amount of testosterone in your body (surgical castration).
- •
- Take TALZENNA with or without food.
- •
- Swallow TALZENNA capsules whole. Do not dissolve or open TALZENNA capsules.
- •
- Your healthcare provider may change your dose of TALZENNA or tell you to stop taking TALZENNA depending on how you respond to treatment.
- •
- If you miss a dose of TALZENNA or vomit, take your next dose at your regular time. Do not take an extra dose to make up for a missed dose.
- •
- If you take too much TALZENNA, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of TALZENNA?
What are the possible side effects of TALZENNA?
TALZENNA may cause serious side effects, including:
The most common side effects of TALZENNA when taken alone include:
- •
- low red blood cell counts
- •
- low white blood cell counts
- •
- low platelet counts
- •
- tiredness or weakness
- •
- increased blood glucose levels
- •
- increased liver function tests
- •
- low calcium in the blood
- •
- nausea
- •
- headache
- •
- vomiting
- •
- hair loss
- •
- diarrhea
- •
- decreased appetite
The most common side effects of TALZENNA when taken in combination with enzalutamide include:
- •
- low red blood cell counts
- •
- low white blood cell counts
- •
- tiredness or weakness
- •
- low platelet counts
- •
- low calcium in the blood
- •
- nausea
- •
- decreased appetite
- •
- low sodium in the blood
- •
- low phosphate in the blood
- •
- bone injuries
- •
- low magnesium in the blood
- •
- dizziness
- •
- increased bilirubin in the blood
- •
- low potassium in the blood
- •
- changes in your sense of taste
TALZENNA may cause fertility problems in males. This may affect your ability to father a child. Talk to your healthcare provider if this is a concern for you.
These are not all of the possible side effects of TALZENNA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store TALZENNA?
How should I store TALZENNA?
- •
- Store TALZENNA at 68°F to 77°F (20°C to 25°C).
Keep TALZENNA and all medicines out of the reach of children.
General information about the safe and effective use of TALZENNA.
General information about the safe and effective use of TALZENNA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use TALZENNA for a condition for which it is not prescribed. Do not give TALZENNA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about TALZENNA that is written for health professionals.
What are the ingredients in TALZENNA?
What are the ingredients in TALZENNA?
Active ingredient:Inactive ingredients:
For more information, go to www.pfizer.com or call 1-800-438-1985.
LAB-1272-5.0
Full Patient Information
Full Patient Information
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
- •
- MDS/AML: Advise patients to contact their healthcare provider if they experience weakness, feeling tired, fever, weight loss, frequent infections, bruising, bleeding easily, breathlessness, blood in urine or stool, and/or laboratory findings of low blood cell counts, or a need for blood transfusions. This may be a sign of hematological toxicity or a more serious uncommon bone marrow problem called MDS or AML, which have been reported in patients who received PARP inhibitors [see Warnings and Precautions (5.1)].
- •
- Myelosuppression: Advise patients that TALZENNA may affect hematopoiesis and can cause anemia, leukopenia/neutropenia, and/or thrombocytopenia [see Warnings and Precautions (5.2)].
- •
- Administration Instructions: Advise patients that TALZENNA can be taken once daily with or without food. Instruct patients that if they miss a dose of TALZENNA, they should take their next normal dose at the usual time. Also advise patients to swallow each capsule whole, and that capsules must not be opened or dissolved [see Dosage and Administration (2.4)].
- •
- Embryo-Fetal Toxicity: Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with TALZENNA and for 7 months after the last dose. Advise male patients with female partners of reproductive potential or who are pregnant to use effective contraception during treatment and for 4 months after receiving the last dose of TALZENNA [see Warnings and Precautions (5.3), Use in Specific Populations (8.1, 8.3)].
- •
- Lactation: Advise patients not to breastfeed while taking TALZENNA and for 1 month after receiving the last dose [see Use in Specific Populations (8.2)].
| This Patient Information has been approved by the U.S. Food and Drug Administration Revised: 2/2024 | |
PATIENT INFORMATION | |
What is the most important information I should know about TALZENNA? Bone marrow problems called Myelodysplastic Syndrome (MDS) or Acute Myeloid Leukemia (AML). Some people who have cancer and who have received previous treatment with chemotherapy or certain other medicines for their cancer have developed MDS or AML during or after treatment with TALZENNA. MDS or AML may lead to death. If you develop MDS or AML, your healthcare provider will stop treatment with TALZENNA. | |
|
|
Your healthcare provider will do blood tests to check your blood cell counts:
See "What are the possible side effects of TALZENNA?" below for other side effects of TALZENNA. | |
What is TALZENNA?
Your healthcare provider will perform a test to make sure that TALZENNA is right for you. It is not known if TALZENNA is safe and effective in children. | |
Before taking TALZENNA, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription medicines, over-the-counter medicines, vitamins, and herbal supplements. Taking TALZENNA and certain other medicines can affect how TALZENNA works and may cause side effects. | |
How should I take TALZENNA?
| |
What are the possible side effects of TALZENNA? TALZENNA may cause serious side effects, including: The most common side effects of TALZENNA when taken alone include: | |
|
|
The most common side effects of TALZENNA when taken in combination with enzalutamide include: | |
|
|
TALZENNA may cause fertility problems in males. This may affect your ability to father a child. Talk to your healthcare provider if this is a concern for you. These are not all of the possible side effects of TALZENNA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |
How should I store TALZENNA?
Keep TALZENNA and all medicines out of the reach of children. | |
General information about the safe and effective use of TALZENNA. | |
What are the ingredients in TALZENNA?
| |
Resources
Didn’t find what you were looking for?
Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5Pm ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this product, click the link below to submit your information: Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer product, please call Pfizer Medical Information at (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800)-332-1088.