PRISTIQ® Clinical Pharmacology
(desvenlafaxine succinate)
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The exact mechanism of the antidepressant action of desvenlafaxine is unknown, but is thought to be related to the potentiation of serotonin and norepinephrine in the central nervous system, through inhibition of their reuptake. Non-clinical studies have shown that desvenlafaxine is a potent and selective SNRI.
12.2 Pharmacodynamics
Desvenlafaxine lacked significant affinity for numerous receptors, including muscarinic-cholinergic, H1-histaminergic, or α1-adrenergic receptors in vitro. Desvenlafaxine also lacked monoamine oxidase (MAO) inhibitory activity.
ECG changes
Electrocardiograms were obtained from 1,492 desvenlafaxine treated patients with major depressive disorder and 984 placebo-treated patients in clinical studies lasting up to 8 weeks. No clinically relevant differences were observed between desvenlafaxine treated and placebo-treated patients for QT, QTc, PR, and QRS intervals. In a thorough QTc study with prospectively determined criteria, desvenlafaxine did not cause QT prolongation. No difference was observed between placebo and desvenlafaxine treatments for the QRS interval.
12.3 Pharmacokinetics
The single-dose pharmacokinetics of desvenlafaxine are linear and dose-proportional in a dose range of 50 to 600 mg (1 to 12 times the recommended approved dosage) per day. With once-daily dosing, steady-state plasma concentrations are achieved within approximately 4 to 5 days. At steady-state, multiple-dose accumulation of desvenlafaxine is linear and predictable from the single-dose pharmacokinetic profile.
Absorption
The absolute oral bioavailability of PRISTIQ after oral administration is about 80%.
Effect of Food
Ingestion of a high-fat meal (800 to 1000 calories) increased desvenlafaxine Cmax about 16% and had no effect on AUC.
Distribution
Steady-state volume of distribution of desvenlafaxine is 3.4 L/kg. Plasma protein binding of desvenlafaxine is 30% and is independent of drug concentration.
Elimination
Metabolism
Desvenlafaxine is primarily metabolized by conjugation (mediated by UGT isoforms) and, to a minor extent, through oxidative metabolism. CYP3A4 mediates the oxidative metabolism (N-demethylation) of desvenlafaxine. The CYP2D6 metabolic pathway is not involved. The pharmacokinetics of desvenlafaxine was similar in subjects with CYP2D6 poor and extensive metabolizer phenotype.
Excretion
Approximately 45% of desvenlafaxine is excreted unchanged in urine at 72 hours after oral administration. Approximately 19% of the administered dose is excreted as the glucuronide metabolite and <5% as the oxidative metabolite (N,O-didesmethylvenlafaxine) in urine.
Specific Populations
No clinically significant differences in the exposures of desvenlafaxine were observed based on ethnicity (White, Black, Hispanic).
The effect of intrinsic patient factors on the pharmacokinetics of desvenlafaxine is presented in Figure 1.
Figure 1 Impact of Intrinsic Factors (Renal, Hepatic Impairment and Population Description) on Desvenlafaxine Pharmacokinetics |
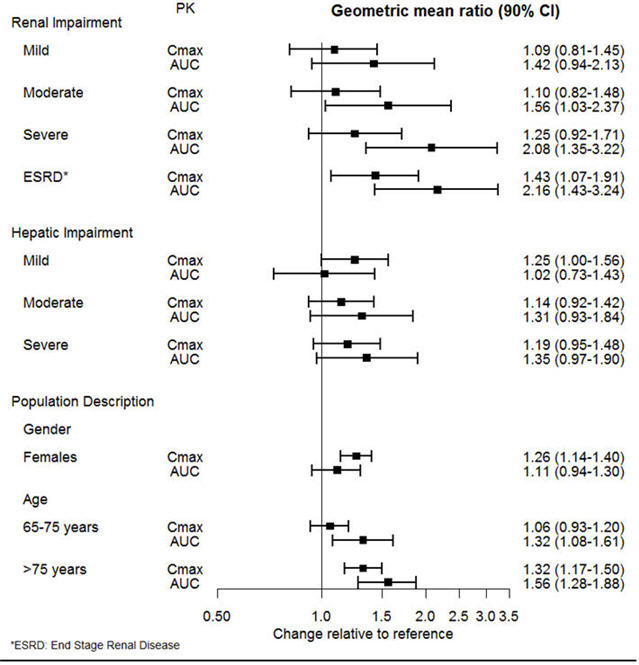 |
Drug Interaction Studies
Clinical Studies
Other Drugs on PRISTIQ
The effect of ketoconazole on the exposures of desvenlafaxine is summarized in Figure 2.
Figure 2. Effect of Other Drugs on Desvenlafaxine Pharmacokinetics |
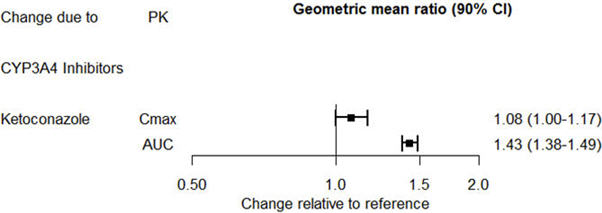 |
PRISTIQ on Other Drugs
The effects of PRISTIQ on the exposures of other drugs are summarized in Figure 3.
Figure 3. Effects of PRISTIQ on Pharmacokinetics of Other Drugs |
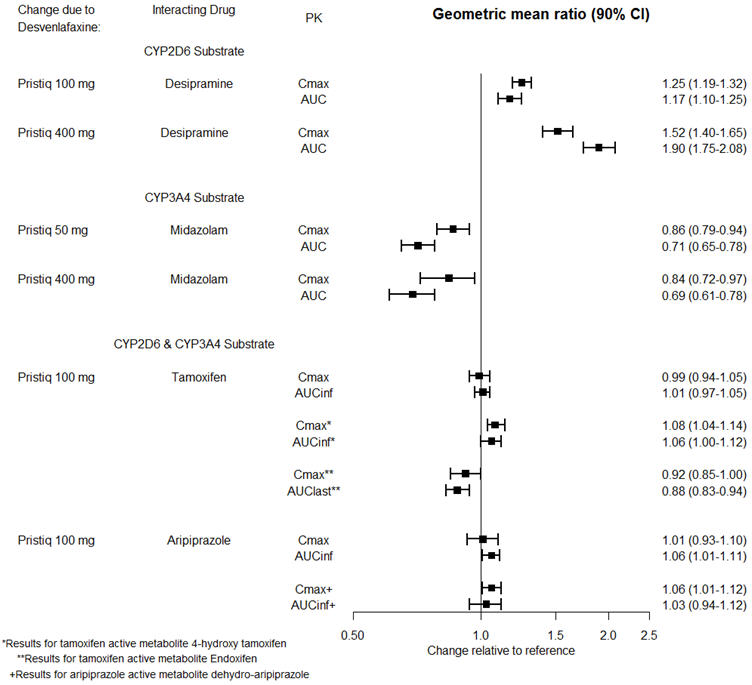 |
In Vitro Studies
Based on in vitro data, drugs that inhibit CYP isozymes 1A1, 1A2, 2A6, 2D6, 2C8, 2C9, 2C19, and 2E1 are not expected to have significant impact on the pharmacokinetic profile of desvenlafaxine.
Desvenlafaxine does not inhibit CYP1A2, 2A6, 2C8, 2C9, 2C19 CYP2D6, or CYP3A4 isozymes. Desvenlafaxine does not induce CYP3A4 either.
Desvenlafaxine is not a substrate or an inhibitor for the P-glycoprotein (P-gp) transporter.
Find PRISTIQ® medical information:
Find PRISTIQ® medical information:
PRISTIQ® Quick Finder
Health Professional Information
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The exact mechanism of the antidepressant action of desvenlafaxine is unknown, but is thought to be related to the potentiation of serotonin and norepinephrine in the central nervous system, through inhibition of their reuptake. Non-clinical studies have shown that desvenlafaxine is a potent and selective SNRI.
12.2 Pharmacodynamics
Desvenlafaxine lacked significant affinity for numerous receptors, including muscarinic-cholinergic, H1-histaminergic, or α1-adrenergic receptors in vitro. Desvenlafaxine also lacked monoamine oxidase (MAO) inhibitory activity.
ECG changes
Electrocardiograms were obtained from 1,492 desvenlafaxine treated patients with major depressive disorder and 984 placebo-treated patients in clinical studies lasting up to 8 weeks. No clinically relevant differences were observed between desvenlafaxine treated and placebo-treated patients for QT, QTc, PR, and QRS intervals. In a thorough QTc study with prospectively determined criteria, desvenlafaxine did not cause QT prolongation. No difference was observed between placebo and desvenlafaxine treatments for the QRS interval.
12.3 Pharmacokinetics
The single-dose pharmacokinetics of desvenlafaxine are linear and dose-proportional in a dose range of 50 to 600 mg (1 to 12 times the recommended approved dosage) per day. With once-daily dosing, steady-state plasma concentrations are achieved within approximately 4 to 5 days. At steady-state, multiple-dose accumulation of desvenlafaxine is linear and predictable from the single-dose pharmacokinetic profile.
Absorption
The absolute oral bioavailability of PRISTIQ after oral administration is about 80%.
Effect of Food
Ingestion of a high-fat meal (800 to 1000 calories) increased desvenlafaxine Cmax about 16% and had no effect on AUC.
Distribution
Steady-state volume of distribution of desvenlafaxine is 3.4 L/kg. Plasma protein binding of desvenlafaxine is 30% and is independent of drug concentration.
Elimination
Metabolism
Desvenlafaxine is primarily metabolized by conjugation (mediated by UGT isoforms) and, to a minor extent, through oxidative metabolism. CYP3A4 mediates the oxidative metabolism (N-demethylation) of desvenlafaxine. The CYP2D6 metabolic pathway is not involved. The pharmacokinetics of desvenlafaxine was similar in subjects with CYP2D6 poor and extensive metabolizer phenotype.
Excretion
Approximately 45% of desvenlafaxine is excreted unchanged in urine at 72 hours after oral administration. Approximately 19% of the administered dose is excreted as the glucuronide metabolite and <5% as the oxidative metabolite (N,O-didesmethylvenlafaxine) in urine.
Specific Populations
No clinically significant differences in the exposures of desvenlafaxine were observed based on ethnicity (White, Black, Hispanic).
The effect of intrinsic patient factors on the pharmacokinetics of desvenlafaxine is presented in Figure 1.
Figure 1 Impact of Intrinsic Factors (Renal, Hepatic Impairment and Population Description) on Desvenlafaxine Pharmacokinetics |
|
Drug Interaction Studies
Clinical Studies
Other Drugs on PRISTIQ
The effect of ketoconazole on the exposures of desvenlafaxine is summarized in Figure 2.
Figure 2. Effect of Other Drugs on Desvenlafaxine Pharmacokinetics |
|
PRISTIQ on Other Drugs
The effects of PRISTIQ on the exposures of other drugs are summarized in Figure 3.
Figure 3. Effects of PRISTIQ on Pharmacokinetics of Other Drugs |
|
In Vitro Studies
Based on in vitro data, drugs that inhibit CYP isozymes 1A1, 1A2, 2A6, 2D6, 2C8, 2C9, 2C19, and 2E1 are not expected to have significant impact on the pharmacokinetic profile of desvenlafaxine.
Desvenlafaxine does not inhibit CYP1A2, 2A6, 2C8, 2C9, 2C19 CYP2D6, or CYP3A4 isozymes. Desvenlafaxine does not induce CYP3A4 either.
Desvenlafaxine is not a substrate or an inhibitor for the P-glycoprotein (P-gp) transporter.
Health Professional Information
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5Pm ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.