linezolid injection in sodium chloride Clinical Pharmacology
()
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
In a randomized, positive- and placebo-controlled crossover thorough QT study, 40 healthy subjects were administered a single linezolid 600 mg dose via a 1 hour IV infusion, a single linezolid 1,200 mg dose via a 1 hour IV infusion, placebo, and a single oral dose of positive control. At both the 600 mg and 1,200 mg linezolid doses, no significant effect on QTc interval was detected at peak plasma concentration or at any other time.
12.3 Pharmacokinetics
The mean pharmacokinetic parameters of linezolid in adults after single and multiple oral and intravenous doses are summarized in Table 8. Plasma concentrations of linezolid at steady-state after oral doses of 600 mg given every 12 hours are shown in Figure 1.
| Dose of Linezolid | Cmax mcg/mL | Cmin mcg/mL | Tmax hrs | AUC* mcg∙h/mL | t1/2 hrs | CL mL/min |
|---|---|---|---|---|---|---|
| ||||||
400 mg tablet | ||||||
single dose† | 8.10 | --- | 1.52 | 55.10 | 5.20 | 146 |
every 12 hours | 11.00 | 3.08 | 1.12 | 73.40 | 4.69 | 110 |
600 mg tablet | ||||||
single dose | 12.70 | --- | 1.28 | 91.40 | 4.26 | 127 |
every 12 hours | 21.20 | 6.15 | 1.03 | 138.00 | 5.40 | 80 |
600 mg IV injection‡ | ||||||
single dose | 12.90 | --- | 0.50 | 80.20 | 4.40 | 138 |
every 12 hours | 15.10 | 3.68 | 0.51 | 89.70 | 4.80 | 123 |
600 mg oral suspension | ||||||
single dose | 11.00 | --- | 0.97 | 80.80 | 4.60 | 141 |
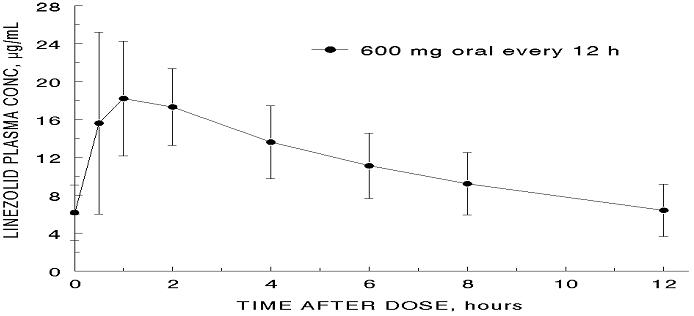
Figure 1. Plasma Concentrations of Linezolid in Adults at Steady-State Following Oral Dosing Every 12 Hours (Mean ± Standard Deviation, n=16)
Absorption
Linezolid is extensively absorbed after oral dosing. Maximum plasma concentrations are reached approximately 1 to 2 hours after dosing, and the absolute bioavailability is approximately 100%. Therefore, linezolid may be given orally or intravenously without dose adjustment.
Linezolid may be administered without regard to the timing of meals. The time to reach the maximum concentration is delayed from 1.5 hours to 2.2 hours and Cmax is decreased by about 17% when high fat food is given with linezolid. However, the total exposure measured as AUC0–∞ is similar under both conditions.
Distribution
Animal and human pharmacokinetic studies have demonstrated that linezolid readily distributes to well-perfused tissues. The plasma protein binding of linezolid is approximately 31% and is concentration-independent. The volume of distribution of linezolid at steady-state averaged 40 to 50 liters in healthy adult volunteers.
Linezolid concentrations have been determined in various fluids from a limited number of subjects in Phase 1 volunteer studies following multiple dosing of linezolid. The ratio of linezolid in saliva relative to plasma was 1.2 to 1 and the ratio of linezolid in sweat relative to plasma was 0.55 to 1.
Metabolism
Linezolid is primarily metabolized by oxidation of the morpholine ring, which results in two inactive ring-opened carboxylic acid metabolites: the aminoethoxyacetic acid metabolite (A), and the hydroxyethyl glycine metabolite (B). Formation of metabolite A is presumed to be formed via an enzymatic pathway whereas metabolite B is mediated by a non-enzymatic chemical oxidation mechanism in vitro. In vitro studies have demonstrated that linezolid is minimally metabolized and may be mediated by human cytochrome P450. However, the metabolic pathway of linezolid is not fully understood.
Excretion
Nonrenal clearance accounts for approximately 65% of the total clearance of linezolid. Under steady-state conditions, approximately 30% of the dose appears in the urine as linezolid, 40% as metabolite B, and 10% as metabolite A. The mean renal clearance of linezolid is 40 mL/min which suggests net tubular reabsorption. Virtually no linezolid appears in the feces, while approximately 6% of the dose appears in the feces as metabolite B, and 3% as metabolite A.
A small degree of nonlinearity in clearance was observed with increasing doses of linezolid, which appears to be due to lower renal and nonrenal clearance of linezolid at higher concentrations. However, the difference in clearance was small and was not reflected in the apparent elimination half-life.
Specific Populations
Geriatric Patients
The pharmacokinetics of linezolid are not significantly altered in elderly patients (65 years or older). Therefore, dose adjustment for geriatric patients is not necessary.
Pediatric Patients
The pharmacokinetics of linezolid following a single intravenous dose were investigated in pediatric patients ranging in age from birth through 17 years (including premature and full-term neonates), in healthy adolescent subjects ranging in age from 12 through 17 years, and in pediatric patients ranging in age from 1 week through 12 years. The pharmacokinetic parameters of linezolid are summarized in Table 9 for the pediatric populations studied and healthy adult subjects after administration of single intravenous doses.
The Cmax and the volume of distribution (Vss) of linezolid are similar regardless of age in pediatric patients. However, plasma clearance of linezolid varies as a function of age. With the exclusion of pre-term neonates less than one week of age, weight-based clearance is most rapid in the youngest age groups ranging from <1 week old to 11 years, resulting in lower single-dose systemic exposure (AUC) and a shorter half-life as compared with adults. As the age of pediatric patients increases, the weight-based clearance of linezolid gradually decreases, and by adolescence mean clearance values approach those observed for the adult population. There is increased inter-subject variability in linezolid clearance and systemic drug exposure (AUC) across all pediatric age groups as compared with adults.
Similar mean daily AUC values were observed in pediatric patients from birth to 11 years of age dosed every 8 hours relative to adolescents or adults dosed every 12 hours. Therefore, the dosage for pediatric patients up to 11 years of age should be 10 mg/kg every 8 hours. Pediatric patients 12 years and older should receive 600 mg every 12 hours [see Dosage and Administration (2)].
| Age Group | Cmax mcg/mL | Vss L/kg | AUC* mcg∙h/mL | t1/2 Hrs | CL mL/min/kg |
|---|---|---|---|---|---|
| Cmax = Maximum plasma concentration; Vss = Volume of distribution; AUC = Area under concentration-time curve; t1/2 = Apparent elimination half-life; CL = Systemic clearance normalized for body weight | |||||
| |||||
Neonatal Patients | |||||
Pre-term† | 12.7 (30%) | 0.81 (24%) | 108 (47%) | 5.6 (46%) | 2.0 (52%) |
<1 week (N = 9)‡ | [9.6, 22.2] | [0.43, 1.05] | [41, 191] | [2.4, 9.8] | [0.9, 4.0] |
Full-term§ | 11.5 (24%) | 0.78 (20%) | 55 (47%) | 3.0 (55%) | 3.8 (55%) |
<1 week (N = 10) ‡ | [8.0, 18.3] | [0.45, 0.96] | [19, 103] | [1.3, 6.1] | [1.5, 8.8] |
Full-term§ | 12.9 (28%) | 0.66 (29%) | 34 (21%) | 1.5 (17%) | 5.1 (22%) |
≥1 week to ≤28 days (N = 10)‡ | [7.7, 21.6] | [0.35, 1.06] | [23, 50] | [1.2, 1.9] | [3.3, 7.2] |
Infant Patients | |||||
>28 days to <3 Months | 11.0 (27%) | 0.79 (26%) | 33 (26%) | 1.8 (28%) | 5.4 (32%) |
(N = 12)‡ | [7.2, 18.0] | [0.42, 1.08] | [17, 48] | [1.2, 2.8] | [3.5, 9.9] |
Pediatric Patients | |||||
3 months through 11 years‡ | 15.1 (30%) | 0.69 (28%) | 58 (54%) | 2.9 (53%) | 3.8 (53%) |
(N = 59) | [6.8, 36.7] | [0.31, 1.50] | [19, 153] | [0.9, 8.0] | [1.0, 8.5] |
Adolescent Subjects and Patients | |||||
12 through 17 years¶ | 16.7 (24%) | 0.61 (15%) | 95 (44%) | 4.1 (46%) | 2.1 (53%) |
(N = 36) | [9.9, 28.9] | [0.44, 0.79] | [32, 178] | [1.3, 8.1] | [0.9, 5.2] |
Adult Subjects# | [8.2, 19.3] | [0.45, 0.84] | [53, 155] | [1.8, 8.3] | [0.9, 3.3] |
(N = 29) | 12.5 (21%) | 0.65 (16%) | 91 (33%) | 4.9 (35%) | 1.7 (34%) |
Gender
Females have a slightly lower volume of distribution of linezolid than males. Plasma concentrations are higher in females than in males, which is partly due to body weight differences. After a 600-mg dose, mean oral clearance is approximately 38% lower in females than in males. However, there are no significant gender differences in mean apparent elimination-rate constant or half-life. Thus, drug exposure in females is not expected to substantially increase beyond levels known to be well tolerated. Therefore, dose adjustment by gender does not appear to be necessary.
Renal Impairment
The pharmacokinetics of the parent drug, linezolid, are not altered in patients with any degree of renal impairment; however, the two primary metabolites of linezolid accumulate in patients with renal impairment, with the amount of accumulation increasing with the severity of renal dysfunction (see Table 10). The pharmacokinetics of linezolid and its two metabolites have also been studied in patients with end-stage renal disease (ESRD) receiving hemodialysis. In the ESRD study, 14 patients were dosed with linezolid 600 mg every 12 hours for 14.5 days (see Table 11). Because similar plasma concentrations of linezolid are achieved regardless of renal function, no dose adjustment is recommended for patients with renal impairment. However, given the absence of information on the clinical significance of accumulation of the primary metabolites, use of linezolid in patients with renal impairment should be weighed against the potential risks of accumulation of these metabolites. Both linezolid and the two metabolites are eliminated by hemodialysis. No information is available on the effect of peritoneal dialysis on the pharmacokinetics of linezolid. Approximately 30% of a dose was eliminated in a 3-hour hemodialysis session beginning 3 hours after the dose of linezolid was administered; therefore, linezolid should be given after hemodialysis.
| Parameter | Healthy Subjects CLCR >80 mL/min | Moderate Renal Impairment 30< CLCR <80 mL/min | Severe Renal Impairment 10< CLCR <30 mL/min |
|---|---|---|---|
| |||
LINEZOLID | |||
AUC0–∞, mcg h/mL | 110 (22) | 128 (53) | 127 (66) |
t1/2, hours | 6.4 (2.2) | 6.1 (1.7) | 7.1 (3.7) |
METABOLITE A | |||
AUC0–48, mcg h/mL | 7.6 (1.9) | 11.7 (4.3) | 56.5 (30.6) |
t1/2, hours | 6.3 (2.1) | 6.6 (2.3) | 9.0 (4.6) |
METABOLITE B* | |||
AUC0–48, mcg h/mL | 30.5 (6.2) | 51.1 (38.5) | 203 (92) |
t1/2, hours | 6.6 (2.7) | 9.9 (7.4) | 11.0 (3.9) |
| Parameter | ESRD Subjects* |
|---|---|
LINEZOLID | |
AUC0–12, mcg h/mL (after last dose) | 181 (52.3) |
t1/2, h (after last dose) | 8.3 (2.4) |
METABOLITE A | |
AUC0–12, mcg h/mL (after last dose) | 153 (40.6) |
t1/2, h (after last dose) | 15.9 (8.5) |
METABOLITE B† | |
AUC0–12, mcg h/mL (after last dose) | 356 (99.7) |
t1/2, h (after last dose) | 34.8 (23.1) |
Hepatic Impairment
The pharmacokinetics of linezolid are not altered in patients (n = 7) with mild-to-moderate hepatic impairment (Child-Pugh class A or B). On the basis of the available information, no dose adjustment is recommended for patients with mild-to-moderate hepatic impairment. The pharmacokinetics of linezolid in patients with severe hepatic impairment have not been evaluated.
Drug Interactions
Drugs Metabolized by Cytochrome P450
Linezolid is not an inducer of cytochrome P450 (CYP450) in rats. In addition, linezolid does not inhibit the activities of clinically significant human CYP isoforms (e.g., 1A2, 2C9, 2C19, 2D6, 2E1, 3A4). Therefore, linezolid is not expected to affect the pharmacokinetics of other drugs metabolized by these major enzymes. Concurrent administration of linezolid does not substantially alter the pharmacokinetic characteristics of (S)-warfarin, which is extensively metabolized by CYP2C9. Drugs such as warfarin and phenytoin, which are CYP2C9 substrates, may be given with linezolid without changes in dosage regimen.
Antibacterial Drugs
Aztreonam: The pharmacokinetics of linezolid or aztreonam are not altered when administered together.
Gentamicin: The pharmacokinetics of linezolid or gentamicin are not altered when administered together.
Antioxidants
The potential for drug-drug interactions with linezolid and the antioxidants Vitamin C and Vitamin E was studied in healthy volunteers. Subjects were administered a 600 mg oral dose of linezolid on Day 1, and another 600 mg dose of linezolid on Day 8. On Days 2–9, subjects were given either Vitamin C (1,000 mg/day) or Vitamin E (800 IU/ day). The AUC0–∞ of linezolid increased 2.3% when co-administered with Vitamin C and 10.9% when co-administered with Vitamin E. No linezolid dose adjustment is recommended during co-administration with Vitamin C or Vitamin E.
Strong CYP 3A4 Inducers
Rifampin: The effect of rifampin on the pharmacokinetics of linezolid was evaluated in a study of 16 healthy adult males. Volunteers were administered oral linezolid 600 mg twice daily for 5 doses with and without rifampin 600 mg once daily for 8 days. Co-administration of rifampin with linezolid resulted in a 21% decrease in linezolid Cmax [90% CI, 15% – 27%] and a 32% decrease in linezolid AUC0–12 [90% CI, 27% – 37%]. The clinical significance of this interaction is unknown. The mechanism of this interaction is not fully understood and may be related to the induction of hepatic enzymes. Other strong inducers of hepatic enzymes (e.g. carbamazepine, phenytoin, phenobarbital) could cause a similar or smaller decrease in linezolid exposure.
Monoamine Oxidase Inhibition
Linezolid is a reversible, nonselective inhibitor of monoamine oxidase. Therefore, linezolid has the potential for interaction with adrenergic and serotonergic agents.
Adrenergic Agents
Some individuals receiving linezolid may experience a reversible enhancement of the pressor response to indirect-acting sympathomimetic agents, vasopressor or dopaminergic agents. Commonly used drugs such as phenylpropanolamine and pseudoephedrine have been specifically studied. Initial doses of adrenergic agents, such as dopamine or epinephrine, should be reduced and titrated to achieve the desired response.
Tyramine: A significant pressor response has been observed in normal adult subjects receiving linezolid and tyramine doses of more than 100 mg. Therefore, patients receiving linezolid need to avoid consuming large amounts of foods or beverages with high tyramine content [see Patient Counseling Information (17)].
Pseudoephedrine HCl or phenylpropanolamine HCl: A reversible enhancement of the pressor response of either pseudoephedrine HCl (PSE) or phenylpropanolamine HCl (PPA) is observed when linezolid is administered to healthy normotensive subjects [see Warnings and Precautions (5.6) and Drug Interactions (7)]. A similar study has not been conducted in hypertensive patients. The interaction studies conducted in normotensive subjects evaluated the blood pressure and heart rate effects of placebo, PPA or PSE alone, linezolid alone, and the combination of steady-state linezolid (600 mg every 12 hours for 3 days) with two doses of PPA (25 mg) or PSE (60 mg) given 4 hours apart. Heart rate was not affected by any of the treatments. Blood pressure was increased with both combination treatments. Maximum blood pressure levels were seen 2 to 3 hours after the second dose of PPA or PSE, and returned to baseline 2 to 3 hours after peak. The results of the PPA study follow, showing the mean (and range) maximum systolic blood pressure in mm Hg: placebo = 121 (103 to 158); linezolid alone = 120 (107 to 135); PPA alone = 125 (106 to 139); PPA with linezolid = 147 (129 to 176). The results from the PSE study were similar to those in the PPA study. The mean maximum increase in systolic blood pressure over baseline was 32 mm Hg (range: 20–52 mm Hg) and 38 mm Hg (range: 18–79 mm Hg) during co-administration of linezolid with pseudoephedrine or phenylpropanolamine, respectively.
Serotonergic Agents
Dextromethorphan: The potential drug-drug interaction with dextromethorphan was studied in healthy volunteers. Subjects were administered dextromethorphan (two 20-mg doses given 4 hours apart) with or without linezolid.
No serotonin syndrome effects (confusion, delirium, restlessness, tremors, blushing, diaphoresis, hyperpyrexia) have been observed in normal subjects receiving linezolid and dextromethorphan.
12.4 Microbiology
Mechanism of Action
Linezolid is a synthetic antibacterial agent of the oxazolidinone class, which has clinical utility in the treatment of infections caused by aerobic Gram-positive bacteria. The in vitro spectrum of activity of linezolid also includes certain Gram-negative bacteria and anaerobic bacteria. Linezolid binds to a site on the bacterial 23S ribosomal RNA of the 50S subunit and prevents the formation of a functional 70S initiation complex, which is essential for bacterial reproduction. The results of time-kill studies have shown linezolid to be bacteriostatic against enterococci and staphylococci. For streptococci, linezolid was found to be bactericidal for the majority of isolates.
Resistance
In vitro studies have shown that point mutations in the 23S rRNA are associated with linezolid resistance. Reports of vancomycin-resistant Enterococcus faecium becoming resistant to linezolid during its clinical use have been published. There are reports of Staphylococcus aureus (methicillin-resistant) developing resistance to linezolid during clinical use. The linezolid resistance in these organisms is associated with a point mutation in the 23S rRNA (substitution of thymine for guanine at position 2576) of the organism. Organisms resistant to oxazolidinones via mutations in chromosomal genes encoding 23S rRNA or ribosomal proteins (L3 and L4) are generally cross-resistant to linezolid. Also linezolid resistance in staphylococci mediated by the enzyme methyltransferase has been reported. This resistance is mediated by the cfr (chloramphenicol-florfenicol) gene located on a plasmid which is transferable between staphylococci.
Interaction with Other Antimicrobial Drugs
In vitro studies have demonstrated additivity or indifference between linezolid and vancomycin, gentamicin, rifampin, imipenem-cilastatin, aztreonam, ampicillin, or streptomycin.
Linezolid has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1)].
Gram-positive bacteria
Enterococcus faecium (vancomycin-resistant isolates only)
Staphylococcus aureus (including methicillin-resistant isolates)
Streptococcus agalactiae
Streptococcus pneumoniae
Streptococcus pyogenes
The following in vitro data are available, but their clinical significance is unknown. Greater than 90% of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the linezolid-susceptible breakpoint for organisms of similar genus. The safety and effectiveness of linezolid in treating clinical infections due to these bacteria have not been established in adequate and well-controlled clinical trials.
Gram-positive bacteria
Enterococcus faecalis (including vancomycin-resistant isolates)
Enterococcus faecium (vancomycin-susceptible isolates)
Staphylococcus epidermidis (including methicillin-resistant isolates)
Staphylococcus haemolyticus
Viridans group streptococci
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
Find linezolid injection in sodium chloride medical information:
Find linezolid injection in sodium chloride medical information:
linezolid injection in sodium chloride Quick Finder
Health Professional Information
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
In a randomized, positive- and placebo-controlled crossover thorough QT study, 40 healthy subjects were administered a single linezolid 600 mg dose via a 1 hour IV infusion, a single linezolid 1,200 mg dose via a 1 hour IV infusion, placebo, and a single oral dose of positive control. At both the 600 mg and 1,200 mg linezolid doses, no significant effect on QTc interval was detected at peak plasma concentration or at any other time.
12.3 Pharmacokinetics
The mean pharmacokinetic parameters of linezolid in adults after single and multiple oral and intravenous doses are summarized in Table 8. Plasma concentrations of linezolid at steady-state after oral doses of 600 mg given every 12 hours are shown in Figure 1.
| Dose of Linezolid | Cmax mcg/mL | Cmin mcg/mL | Tmax hrs | AUC* mcg∙h/mL | t1/2 hrs | CL mL/min |
|---|---|---|---|---|---|---|
| ||||||
400 mg tablet | ||||||
single dose† | 8.10 | --- | 1.52 | 55.10 | 5.20 | 146 |
every 12 hours | 11.00 | 3.08 | 1.12 | 73.40 | 4.69 | 110 |
600 mg tablet | ||||||
single dose | 12.70 | --- | 1.28 | 91.40 | 4.26 | 127 |
every 12 hours | 21.20 | 6.15 | 1.03 | 138.00 | 5.40 | 80 |
600 mg IV injection‡ | ||||||
single dose | 12.90 | --- | 0.50 | 80.20 | 4.40 | 138 |
every 12 hours | 15.10 | 3.68 | 0.51 | 89.70 | 4.80 | 123 |
600 mg oral suspension | ||||||
single dose | 11.00 | --- | 0.97 | 80.80 | 4.60 | 141 |
Figure 1. Plasma Concentrations of Linezolid in Adults at Steady-State Following Oral Dosing Every 12 Hours (Mean ± Standard Deviation, n=16)
Absorption
Linezolid is extensively absorbed after oral dosing. Maximum plasma concentrations are reached approximately 1 to 2 hours after dosing, and the absolute bioavailability is approximately 100%. Therefore, linezolid may be given orally or intravenously without dose adjustment.
Linezolid may be administered without regard to the timing of meals. The time to reach the maximum concentration is delayed from 1.5 hours to 2.2 hours and Cmax is decreased by about 17% when high fat food is given with linezolid. However, the total exposure measured as AUC0–∞ is similar under both conditions.
Distribution
Animal and human pharmacokinetic studies have demonstrated that linezolid readily distributes to well-perfused tissues. The plasma protein binding of linezolid is approximately 31% and is concentration-independent. The volume of distribution of linezolid at steady-state averaged 40 to 50 liters in healthy adult volunteers.
Linezolid concentrations have been determined in various fluids from a limited number of subjects in Phase 1 volunteer studies following multiple dosing of linezolid. The ratio of linezolid in saliva relative to plasma was 1.2 to 1 and the ratio of linezolid in sweat relative to plasma was 0.55 to 1.
Metabolism
Linezolid is primarily metabolized by oxidation of the morpholine ring, which results in two inactive ring-opened carboxylic acid metabolites: the aminoethoxyacetic acid metabolite (A), and the hydroxyethyl glycine metabolite (B). Formation of metabolite A is presumed to be formed via an enzymatic pathway whereas metabolite B is mediated by a non-enzymatic chemical oxidation mechanism in vitro. In vitro studies have demonstrated that linezolid is minimally metabolized and may be mediated by human cytochrome P450. However, the metabolic pathway of linezolid is not fully understood.
Excretion
Nonrenal clearance accounts for approximately 65% of the total clearance of linezolid. Under steady-state conditions, approximately 30% of the dose appears in the urine as linezolid, 40% as metabolite B, and 10% as metabolite A. The mean renal clearance of linezolid is 40 mL/min which suggests net tubular reabsorption. Virtually no linezolid appears in the feces, while approximately 6% of the dose appears in the feces as metabolite B, and 3% as metabolite A.
A small degree of nonlinearity in clearance was observed with increasing doses of linezolid, which appears to be due to lower renal and nonrenal clearance of linezolid at higher concentrations. However, the difference in clearance was small and was not reflected in the apparent elimination half-life.
Specific Populations
Geriatric Patients
The pharmacokinetics of linezolid are not significantly altered in elderly patients (65 years or older). Therefore, dose adjustment for geriatric patients is not necessary.
Pediatric Patients
The pharmacokinetics of linezolid following a single intravenous dose were investigated in pediatric patients ranging in age from birth through 17 years (including premature and full-term neonates), in healthy adolescent subjects ranging in age from 12 through 17 years, and in pediatric patients ranging in age from 1 week through 12 years. The pharmacokinetic parameters of linezolid are summarized in Table 9 for the pediatric populations studied and healthy adult subjects after administration of single intravenous doses.
The Cmax and the volume of distribution (Vss) of linezolid are similar regardless of age in pediatric patients. However, plasma clearance of linezolid varies as a function of age. With the exclusion of pre-term neonates less than one week of age, weight-based clearance is most rapid in the youngest age groups ranging from <1 week old to 11 years, resulting in lower single-dose systemic exposure (AUC) and a shorter half-life as compared with adults. As the age of pediatric patients increases, the weight-based clearance of linezolid gradually decreases, and by adolescence mean clearance values approach those observed for the adult population. There is increased inter-subject variability in linezolid clearance and systemic drug exposure (AUC) across all pediatric age groups as compared with adults.
Similar mean daily AUC values were observed in pediatric patients from birth to 11 years of age dosed every 8 hours relative to adolescents or adults dosed every 12 hours. Therefore, the dosage for pediatric patients up to 11 years of age should be 10 mg/kg every 8 hours. Pediatric patients 12 years and older should receive 600 mg every 12 hours [see Dosage and Administration (2)].
| Age Group | Cmax mcg/mL | Vss L/kg | AUC* mcg∙h/mL | t1/2 Hrs | CL mL/min/kg |
|---|---|---|---|---|---|
| Cmax = Maximum plasma concentration; Vss = Volume of distribution; AUC = Area under concentration-time curve; t1/2 = Apparent elimination half-life; CL = Systemic clearance normalized for body weight | |||||
| |||||
Neonatal Patients | |||||
Pre-term† | 12.7 (30%) | 0.81 (24%) | 108 (47%) | 5.6 (46%) | 2.0 (52%) |
<1 week (N = 9)‡ | [9.6, 22.2] | [0.43, 1.05] | [41, 191] | [2.4, 9.8] | [0.9, 4.0] |
Full-term§ | 11.5 (24%) | 0.78 (20%) | 55 (47%) | 3.0 (55%) | 3.8 (55%) |
<1 week (N = 10) ‡ | [8.0, 18.3] | [0.45, 0.96] | [19, 103] | [1.3, 6.1] | [1.5, 8.8] |
Full-term§ | 12.9 (28%) | 0.66 (29%) | 34 (21%) | 1.5 (17%) | 5.1 (22%) |
≥1 week to ≤28 days (N = 10)‡ | [7.7, 21.6] | [0.35, 1.06] | [23, 50] | [1.2, 1.9] | [3.3, 7.2] |
Infant Patients | |||||
>28 days to <3 Months | 11.0 (27%) | 0.79 (26%) | 33 (26%) | 1.8 (28%) | 5.4 (32%) |
(N = 12)‡ | [7.2, 18.0] | [0.42, 1.08] | [17, 48] | [1.2, 2.8] | [3.5, 9.9] |
Pediatric Patients | |||||
3 months through 11 years‡ | 15.1 (30%) | 0.69 (28%) | 58 (54%) | 2.9 (53%) | 3.8 (53%) |
(N = 59) | [6.8, 36.7] | [0.31, 1.50] | [19, 153] | [0.9, 8.0] | [1.0, 8.5] |
Adolescent Subjects and Patients | |||||
12 through 17 years¶ | 16.7 (24%) | 0.61 (15%) | 95 (44%) | 4.1 (46%) | 2.1 (53%) |
(N = 36) | [9.9, 28.9] | [0.44, 0.79] | [32, 178] | [1.3, 8.1] | [0.9, 5.2] |
Adult Subjects# | [8.2, 19.3] | [0.45, 0.84] | [53, 155] | [1.8, 8.3] | [0.9, 3.3] |
(N = 29) | 12.5 (21%) | 0.65 (16%) | 91 (33%) | 4.9 (35%) | 1.7 (34%) |
Gender
Females have a slightly lower volume of distribution of linezolid than males. Plasma concentrations are higher in females than in males, which is partly due to body weight differences. After a 600-mg dose, mean oral clearance is approximately 38% lower in females than in males. However, there are no significant gender differences in mean apparent elimination-rate constant or half-life. Thus, drug exposure in females is not expected to substantially increase beyond levels known to be well tolerated. Therefore, dose adjustment by gender does not appear to be necessary.
Renal Impairment
The pharmacokinetics of the parent drug, linezolid, are not altered in patients with any degree of renal impairment; however, the two primary metabolites of linezolid accumulate in patients with renal impairment, with the amount of accumulation increasing with the severity of renal dysfunction (see Table 10). The pharmacokinetics of linezolid and its two metabolites have also been studied in patients with end-stage renal disease (ESRD) receiving hemodialysis. In the ESRD study, 14 patients were dosed with linezolid 600 mg every 12 hours for 14.5 days (see Table 11). Because similar plasma concentrations of linezolid are achieved regardless of renal function, no dose adjustment is recommended for patients with renal impairment. However, given the absence of information on the clinical significance of accumulation of the primary metabolites, use of linezolid in patients with renal impairment should be weighed against the potential risks of accumulation of these metabolites. Both linezolid and the two metabolites are eliminated by hemodialysis. No information is available on the effect of peritoneal dialysis on the pharmacokinetics of linezolid. Approximately 30% of a dose was eliminated in a 3-hour hemodialysis session beginning 3 hours after the dose of linezolid was administered; therefore, linezolid should be given after hemodialysis.
| Parameter | Healthy Subjects CLCR >80 mL/min | Moderate Renal Impairment 30< CLCR <80 mL/min | Severe Renal Impairment 10< CLCR <30 mL/min |
|---|---|---|---|
| |||
LINEZOLID | |||
AUC0–∞, mcg h/mL | 110 (22) | 128 (53) | 127 (66) |
t1/2, hours | 6.4 (2.2) | 6.1 (1.7) | 7.1 (3.7) |
METABOLITE A | |||
AUC0–48, mcg h/mL | 7.6 (1.9) | 11.7 (4.3) | 56.5 (30.6) |
t1/2, hours | 6.3 (2.1) | 6.6 (2.3) | 9.0 (4.6) |
METABOLITE B* | |||
AUC0–48, mcg h/mL | 30.5 (6.2) | 51.1 (38.5) | 203 (92) |
t1/2, hours | 6.6 (2.7) | 9.9 (7.4) | 11.0 (3.9) |
| Parameter | ESRD Subjects* |
|---|---|
LINEZOLID | |
AUC0–12, mcg h/mL (after last dose) | 181 (52.3) |
t1/2, h (after last dose) | 8.3 (2.4) |
METABOLITE A | |
AUC0–12, mcg h/mL (after last dose) | 153 (40.6) |
t1/2, h (after last dose) | 15.9 (8.5) |
METABOLITE B† | |
AUC0–12, mcg h/mL (after last dose) | 356 (99.7) |
t1/2, h (after last dose) | 34.8 (23.1) |
Hepatic Impairment
The pharmacokinetics of linezolid are not altered in patients (n = 7) with mild-to-moderate hepatic impairment (Child-Pugh class A or B). On the basis of the available information, no dose adjustment is recommended for patients with mild-to-moderate hepatic impairment. The pharmacokinetics of linezolid in patients with severe hepatic impairment have not been evaluated.
Drug Interactions
Drugs Metabolized by Cytochrome P450
Linezolid is not an inducer of cytochrome P450 (CYP450) in rats. In addition, linezolid does not inhibit the activities of clinically significant human CYP isoforms (e.g., 1A2, 2C9, 2C19, 2D6, 2E1, 3A4). Therefore, linezolid is not expected to affect the pharmacokinetics of other drugs metabolized by these major enzymes. Concurrent administration of linezolid does not substantially alter the pharmacokinetic characteristics of (S)-warfarin, which is extensively metabolized by CYP2C9. Drugs such as warfarin and phenytoin, which are CYP2C9 substrates, may be given with linezolid without changes in dosage regimen.
Antibacterial Drugs
Aztreonam: The pharmacokinetics of linezolid or aztreonam are not altered when administered together.
Gentamicin: The pharmacokinetics of linezolid or gentamicin are not altered when administered together.
Antioxidants
The potential for drug-drug interactions with linezolid and the antioxidants Vitamin C and Vitamin E was studied in healthy volunteers. Subjects were administered a 600 mg oral dose of linezolid on Day 1, and another 600 mg dose of linezolid on Day 8. On Days 2–9, subjects were given either Vitamin C (1,000 mg/day) or Vitamin E (800 IU/ day). The AUC0–∞ of linezolid increased 2.3% when co-administered with Vitamin C and 10.9% when co-administered with Vitamin E. No linezolid dose adjustment is recommended during co-administration with Vitamin C or Vitamin E.
Strong CYP 3A4 Inducers
Rifampin: The effect of rifampin on the pharmacokinetics of linezolid was evaluated in a study of 16 healthy adult males. Volunteers were administered oral linezolid 600 mg twice daily for 5 doses with and without rifampin 600 mg once daily for 8 days. Co-administration of rifampin with linezolid resulted in a 21% decrease in linezolid Cmax [90% CI, 15% – 27%] and a 32% decrease in linezolid AUC0–12 [90% CI, 27% – 37%]. The clinical significance of this interaction is unknown. The mechanism of this interaction is not fully understood and may be related to the induction of hepatic enzymes. Other strong inducers of hepatic enzymes (e.g. carbamazepine, phenytoin, phenobarbital) could cause a similar or smaller decrease in linezolid exposure.
Monoamine Oxidase Inhibition
Linezolid is a reversible, nonselective inhibitor of monoamine oxidase. Therefore, linezolid has the potential for interaction with adrenergic and serotonergic agents.
Adrenergic Agents
Some individuals receiving linezolid may experience a reversible enhancement of the pressor response to indirect-acting sympathomimetic agents, vasopressor or dopaminergic agents. Commonly used drugs such as phenylpropanolamine and pseudoephedrine have been specifically studied. Initial doses of adrenergic agents, such as dopamine or epinephrine, should be reduced and titrated to achieve the desired response.
Tyramine: A significant pressor response has been observed in normal adult subjects receiving linezolid and tyramine doses of more than 100 mg. Therefore, patients receiving linezolid need to avoid consuming large amounts of foods or beverages with high tyramine content [see Patient Counseling Information (17)].
Pseudoephedrine HCl or phenylpropanolamine HCl: A reversible enhancement of the pressor response of either pseudoephedrine HCl (PSE) or phenylpropanolamine HCl (PPA) is observed when linezolid is administered to healthy normotensive subjects [see Warnings and Precautions (5.6) and Drug Interactions (7)]. A similar study has not been conducted in hypertensive patients. The interaction studies conducted in normotensive subjects evaluated the blood pressure and heart rate effects of placebo, PPA or PSE alone, linezolid alone, and the combination of steady-state linezolid (600 mg every 12 hours for 3 days) with two doses of PPA (25 mg) or PSE (60 mg) given 4 hours apart. Heart rate was not affected by any of the treatments. Blood pressure was increased with both combination treatments. Maximum blood pressure levels were seen 2 to 3 hours after the second dose of PPA or PSE, and returned to baseline 2 to 3 hours after peak. The results of the PPA study follow, showing the mean (and range) maximum systolic blood pressure in mm Hg: placebo = 121 (103 to 158); linezolid alone = 120 (107 to 135); PPA alone = 125 (106 to 139); PPA with linezolid = 147 (129 to 176). The results from the PSE study were similar to those in the PPA study. The mean maximum increase in systolic blood pressure over baseline was 32 mm Hg (range: 20–52 mm Hg) and 38 mm Hg (range: 18–79 mm Hg) during co-administration of linezolid with pseudoephedrine or phenylpropanolamine, respectively.
Serotonergic Agents
Dextromethorphan: The potential drug-drug interaction with dextromethorphan was studied in healthy volunteers. Subjects were administered dextromethorphan (two 20-mg doses given 4 hours apart) with or without linezolid.
No serotonin syndrome effects (confusion, delirium, restlessness, tremors, blushing, diaphoresis, hyperpyrexia) have been observed in normal subjects receiving linezolid and dextromethorphan.
12.4 Microbiology
Mechanism of Action
Linezolid is a synthetic antibacterial agent of the oxazolidinone class, which has clinical utility in the treatment of infections caused by aerobic Gram-positive bacteria. The in vitro spectrum of activity of linezolid also includes certain Gram-negative bacteria and anaerobic bacteria. Linezolid binds to a site on the bacterial 23S ribosomal RNA of the 50S subunit and prevents the formation of a functional 70S initiation complex, which is essential for bacterial reproduction. The results of time-kill studies have shown linezolid to be bacteriostatic against enterococci and staphylococci. For streptococci, linezolid was found to be bactericidal for the majority of isolates.
Resistance
In vitro studies have shown that point mutations in the 23S rRNA are associated with linezolid resistance. Reports of vancomycin-resistant Enterococcus faecium becoming resistant to linezolid during its clinical use have been published. There are reports of Staphylococcus aureus (methicillin-resistant) developing resistance to linezolid during clinical use. The linezolid resistance in these organisms is associated with a point mutation in the 23S rRNA (substitution of thymine for guanine at position 2576) of the organism. Organisms resistant to oxazolidinones via mutations in chromosomal genes encoding 23S rRNA or ribosomal proteins (L3 and L4) are generally cross-resistant to linezolid. Also linezolid resistance in staphylococci mediated by the enzyme methyltransferase has been reported. This resistance is mediated by the cfr (chloramphenicol-florfenicol) gene located on a plasmid which is transferable between staphylococci.
Interaction with Other Antimicrobial Drugs
In vitro studies have demonstrated additivity or indifference between linezolid and vancomycin, gentamicin, rifampin, imipenem-cilastatin, aztreonam, ampicillin, or streptomycin.
Linezolid has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1)].
Gram-positive bacteria
Enterococcus faecium (vancomycin-resistant isolates only)
Staphylococcus aureus (including methicillin-resistant isolates)
Streptococcus agalactiae
Streptococcus pneumoniae
Streptococcus pyogenes
The following in vitro data are available, but their clinical significance is unknown. Greater than 90% of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the linezolid-susceptible breakpoint for organisms of similar genus. The safety and effectiveness of linezolid in treating clinical infections due to these bacteria have not been established in adequate and well-controlled clinical trials.
Gram-positive bacteria
Enterococcus faecalis (including vancomycin-resistant isolates)
Enterococcus faecium (vancomycin-susceptible isolates)
Staphylococcus epidermidis (including methicillin-resistant isolates)
Staphylococcus haemolyticus
Viridans group streptococci
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
Health Professional Information
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5Pm ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.