Levofloxacin in 5% Dextrose Injection, USP Clinical Pharmacology
()
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Levofloxacin is a member of the fluoroquinolone class of antibacterial agents [see Microbiology (12.4)].
12.3 Pharmacokinetics
The mean ± SD pharmacokinetic parameters of levofloxacin determined under single and steady-state conditions following oral tablet, oral solution, or intravenous (IV) doses of levofloxacin are summarized in Table 8.
| Regimen | Cmax (mcg/mL) | Tmax (h) | AUC (mcg∙h/mL) | CL/F* (mL/min) | Vd/F† (L) | t1/2 (h) | CLR (mL/min) |
|---|---|---|---|---|---|---|---|
| |||||||
| Single dose | |||||||
| 250 mg oral tablet‡ | 2.8 ± 0.4 | 1.6 ± 1.0 | 27.2 ± 3.9 | 156 ± 20 | ND | 7.3 ± 0.9 | 142 ± 21 |
| 500 mg oral tablet‡§ | 5.1 ± 0.8 | 1.3 ± 0.6 | 47.9 ± 6.8 | 178 ± 28 | ND | 6.3 ± 0.6 | 103 ± 30 |
| 500 mg oral solution¶ | 5.8 ± 1.8 | 0.8 ± 0.7 | 47.8 ± 10.8 | 183 ± 40 | 112 ± 37.2 | 7.0 ± 1.4 | ND |
| 500 mg IV‡ | 6.2 ± 1.0 | 1.0 ± 0.1 | 48.3 ± 5.4 | 175 ± 20 | 90 ± 11 | 6.4 ± 0.7 | 112 ± 25 |
| 750 mg oral tablet#§ | 9.3 ± 1.6 | 1.6 ± 0.8 | 101 ± 20 | 129 ± 24 | 83 ± 17 | 7.5 ± 0.9 | ND |
| 750 mg IV# | 11.5 ± 4.0Þ | ND | 110 ± 40 | 126 ± 39 | 75 ± 13 | 7.5 ± 1.6 | ND |
| Multiple dose | |||||||
| 500 mg every 24h oral tablet‡ | 5.7 ± 1.4 | 1.1 ± 0.4 | 47.5 ± 6.7 | 175 ± 25 | 102 ± 22 | 7.6 ± 1.6 | 116 ± 31 |
| 500 mg every 24h IV‡ | 6.4 ± 0.8 | ND | 54.6 ± 11.1 | 158 ± 29 | 91 ± 12 | 7.0 ± 0.8 | 99 ± 28 |
| 500 mg or 250 mg every 24h IV, patients with bacterial infectionß | 8.7 ± 4.0à | ND | 72.5 ± 51.2à | 154 ± 72 | 111 ± 58 | ND | ND |
| 750 mg every 24h oral tablet# | 8.6 ± 1.9 | 1.4 ± 0.5 | 90.7 ± 17.6 | 143 ± 29 | 100 ± 16 | 8.8 ± 1.5 | 116 ± 28 |
| 750 mg every 24h IV# | 12.1 ± 4.1Þ | ND | 108 ± 34 | 126 ± 37 | 80 ± 27 | 7.9 ± 1.9 | ND |
| 500 mg oral tablet single dose, effects of gender and age: | |||||||
| Maleè | 5.5 ± 1.1 | 1.2 ± 0.4 | 54.4 ± 18.9 | 166 ± 44 | 89 ± 13 | 7.5 ± 2.1 | 126 ± 38 |
| Femaleð | 7.0 ± 1.6 | 1.7 ± 0.5 | 67.7 ± 24.2 | 136 ± 44 | 62 ± 16 | 6.1 ± 0.8 | 106 ± 40 |
| Youngø | 5.5 ± 1.0 | 1.5 ± 0.6 | 47.5 ± 9.8 | 182 ± 35 | 83 ± 18 | 6.0 ± 0.9 | 140 ± 33 |
| Elderlyý | 7.0 ± 1.6 | 1.4 ± 0.5 | 74.7 ± 23.3 | 121 ± 33 | 67 ± 19 | 7.6 ± 2.0 | 91 ± 29 |
| 500 mg oral single dose tablet, patients with renal insufficiency: | |||||||
| CLCR 50–80 mL/min | 7.5 ± 1.8 | 1.5 ± 0.5 | 95.6 ± 11.8 | 88 ± 10 | ND | 9.1 ± 0.9 | 57 ± 8 |
| CLCR 20–49 mL/min | 7.1 ± 3.1 | 2.1 ± 1.3 | 182.1 ± 62.6 | 51 ± 19 | ND | 27 ± 10 | 26 ± 13 |
| CLCR <20 mL/min | 8.2 ± 2.6 | 1.1 ± 1.0 | 263.5 ± 72.5 | 33 ± 8 | ND | 35 ± 5 | 13 ± 3 |
| Hemodialysis | 5.7 ± 1.0 | 2.8 ± 2.2 | ND | ND | ND | 76 ± 42 | ND |
| CAPD | 6.9 ± 2.3 | 1.4 ± 1.1 | ND | ND | ND | 51 ± 24 | ND |
Absorption
Levofloxacin is rapidly and essentially completely absorbed after oral administration. Peak plasma concentrations are usually attained one to two hours after oral dosing. The absolute bioavailability of levofloxacin from a 500 mg tablet and a 750 mg tablet of levofloxacin are both approximately 99%, demonstrating complete oral absorption of levofloxacin. Following a single intravenous dose of levofloxacin to healthy volunteers, the mean ± SD peak plasma concentration attained was 6.2 ± 1.0 mcg/mL after a 500 mg dose infused over 60 minutes and 11.5 ± 4.0 mcg/mL after a 750 mg dose infused over 90 minutes. Levofloxacin Oral Solution and Tablet formulations are bioequivalent.
Levofloxacin pharmacokinetics are linear and predictable after single and multiple oral or IV dosing regimens. Steady-state conditions are reached within 48 hours following a 500 mg or 750 mg once-daily dosage regimen. The mean ± SD peak and trough plasma concentrations attained following multiple once-daily oral dosage regimens were approximately 5.7 ± 1.4 and 0.5 ± 0.2 mcg/mL after the 500 mg doses, and 8.6 ± 1.9 and 1.1 ± 0.4 mcg/mL after the 750 mg doses, respectively. The mean ± SD peak and trough plasma concentrations attained following multiple once-daily IV regimens were approximately 6.4 ± 0.8 and 0.6 ± 0.2 mcg/mL after the 500 mg doses, and 12.1 ± 4.1 and 1.3 ± 0.71 mcg/mL after the 750 mg doses, respectively. Oral administration of a 500 mg dose of levofloxacin with food prolongs the time to peak concentration by approximately 1 hour and decreases the peak concentration by approximately 14% following tablet and approximately 25% following oral solution administration. Therefore, levofloxacin tablets can be administered without regard to food. It is recommended that levofloxacin oral solution be taken 1 hour before or 2 hours after eating.
The plasma concentration profile of levofloxacin after IV administration is similar and comparable in extent of exposure (AUC) to that observed for levofloxacin tablets when equal doses (mg/mg) are administered. Therefore, the oral and IV routes of administration can be considered interchangeable (see Figure 2 and Figure 3).
| Figure 2: Mean Levofloxacin Plasma Concentration vs. Time Profile: 750 mg |
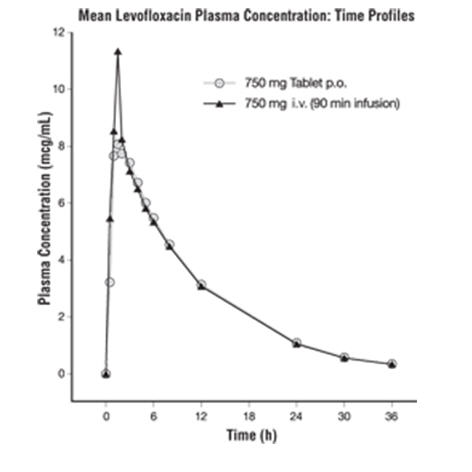 |
| Figure 3: Mean Levofloxacin Plasma Concentration vs. Time Profile: 500 mg |
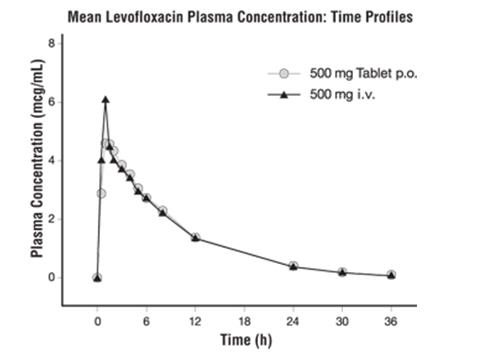 |
Distribution
The mean volume of distribution of levofloxacin generally ranges from 74 to 112 L after single and multiple 500 mg or 750 mg doses, indicating widespread distribution into body tissues. Levofloxacin reaches its peak levels in skin tissues and in blister fluid of healthy subjects at approximately 3 hours after dosing. The skin tissue biopsy to plasma AUC ratio is approximately 2 and the blister fluid to plasma AUC ratio is approximately 1 following multiple once-daily oral administration of 750 mg and 500 mg doses of levofloxacin, respectively, to healthy subjects. Levofloxacin also penetrates well into lung tissues. Lung tissue concentrations were generally 2- to 5-fold higher than plasma concentrations and ranged from approximately 2.4 to 11.3 mcg/g over a 24-hour period after a single 500 mg oral dose.
In vitro, over a clinically relevant range (1 to 10 mcg/mL) of serum/plasma levofloxacin concentrations, levofloxacin is approximately 24 to 38% bound to serum proteins across all species studied, as determined by the equilibrium dialysis method. Levofloxacin is mainly bound to serum albumin in humans. Levofloxacin binding to serum proteins is independent of the drug concentration.
Metabolism
Levofloxacin is stereochemically stable in plasma and urine and does not invert metabolically to its enantiomer, D-ofloxacin. Levofloxacin undergoes limited metabolism in humans and is primarily excreted as unchanged drug in the urine. Following oral administration, approximately 87% of an administered dose was recovered as unchanged drug in urine within 48 hours, whereas less than 4% of the dose was recovered in feces in 72 hours. Less than 5% of an administered dose was recovered in the urine as the desmethyl and N-oxide metabolites, the only metabolites identified in humans. These metabolites have little relevant pharmacological activity.
Excretion
Levofloxacin is excreted largely as unchanged drug in the urine. The mean terminal plasma elimination half-life of levofloxacin ranges from approximately 6 to 8 hours following single or multiple doses of levofloxacin given orally or intravenously. The mean apparent total body clearance and renal clearance range from approximately 144 to 226 mL/min and 96 to 142 mL/min, respectively. Renal clearance in excess of the glomerular filtration rate suggests that tubular secretion of levofloxacin occurs in addition to its glomerular filtration. Concomitant administration of either cimetidine or probenecid results in approximately 24% and 35% reduction in the levofloxacin renal clearance, respectively, indicating that secretion of levofloxacin occurs in the renal proximal tubule. No levofloxacin crystals were found in any of the urine samples freshly collected from subjects receiving levofloxacin.
Geriatric
There are no significant differences in levofloxacin pharmacokinetics between young and elderly subjects when the subjects' differences in creatinine clearance are taken into consideration. Following a 500 mg oral dose of levofloxacin to healthy elderly subjects (66 – 80 years of age), the mean terminal plasma elimination half-life of levofloxacin was about 7.6 hours, as compared to approximately 6 hours in younger adults. The difference was attributable to the variation in renal function status of the subjects and was not believed to be clinically significant. Drug absorption appears to be unaffected by age. Levofloxacin dose adjustment based on age alone is not necessary [see Use in Specific Populations (8.5)].
Pediatrics
The pharmacokinetics of levofloxacin following a single 7 mg/kg intravenous dose were investigated in pediatric patients ranging in age from 6 months to 16 years. Pediatric patients cleared levofloxacin faster than adult patients, resulting in lower plasma exposures than adults for a given mg/kg dose. Subsequent pharmacokinetic analyses predicted that a dosage regimen of 8 mg/kg every 12 hours (not to exceed 250 mg per dose) for pediatric patients 6 months to 17 years of age would achieve comparable steady state plasma exposures (AUC0–24 and Cmax) to those observed in adult patients administered 500 mg of levofloxacin once every 24 hours.
Gender
There are no significant differences in levofloxacin pharmacokinetics between male and female subjects when subjects' differences in creatinine clearance are taken into consideration. Following a 500 mg oral dose of levofloxacin to healthy male subjects, the mean terminal plasma elimination half-life of levofloxacin was about 7.5 hours, as compared to approximately 6.1 hours in female subjects. This difference was attributable to the variation in renal function status of the male and female subjects and was not believed to be clinically significant. Drug absorption appears to be unaffected by the gender of the subjects. Dose adjustment based on gender alone is not necessary.
Race
The effect of race on levofloxacin pharmacokinetics was examined through a covariate analysis performed on data from 72 subjects: 48 white and 24 non-white. The apparent total body clearance and apparent volume of distribution were not affected by the race of the subjects.
Renal Impairment
Clearance of levofloxacin is substantially reduced and plasma elimination half-life is substantially prolonged in adult patients with impaired renal function (creatinine clearance <50 mL/min), requiring dosage adjustment in such patients to avoid accumulation. Neither hemodialysis nor continuous ambulatory peritoneal dialysis (CAPD) is effective in removal of levofloxacin from the body, indicating that supplemental doses of levofloxacin are not required following hemodialysis or CAPD [see Dosage and Administration (2.3), Use in Specific Populations (8.6)].
Hepatic Impairment
Pharmacokinetic studies in hepatically impaired patients have not been conducted. Due to the limited extent of levofloxacin metabolism, the pharmacokinetics of levofloxacin are not expected to be affected by hepatic impairment [see Use in Specific Populations (8.7)].
Bacterial Infection
The pharmacokinetics of levofloxacin in patients with serious community-acquired bacterial infections are comparable to those observed in healthy subjects.
Drug-Drug Interactions
The potential for pharmacokinetic drug interactions between levofloxacin and antacids, warfarin, theophylline, cyclosporine, digoxin, probenecid, and cimetidine has been evaluated [see Drug Interactions (7)].
12.4 Microbiology
Mechanism of Action
Levofloxacin is the L-isomer of the racemate, ofloxacin, a quinolone antimicrobial agent. The antibacterial activity of ofloxacin resides primarily in the L-isomer. The mechanism of action of levofloxacin and other fluoroquinolone antimicrobials involves inhibition of bacterial topoisomerase IV and DNA gyrase (both of which are type II topoisomerases), enzymes required for DNA replication, transcription, repair and recombination.
Resistance
Fluoroquinolone resistance can arise through mutations in defined regions of DNA gyrase or topoisomerase IV, termed the Quinolone-Resistance Determining Regions (QRDRs), or through altered efflux.
Fluoroquinolones, including levofloxacin, differ in chemical structure and mode of action from aminoglycosides, macrolides and β-lactam antibiotics, including penicillins. Fluoroquinolones may, therefore, be active against bacteria resistant to these antimicrobials.
Resistance to levofloxacin due to spontaneous mutation in vitro is a rare occurrence (range: 10-9 to 10-10). Cross-resistance has been observed between levofloxacin and some other fluoroquinolones, some microorganisms resistant to other fluoroquinolones may be susceptible to levofloxacin.
Antimicrobial Activity
Levofloxacin has in vitro activity against Gram-negative and Gram-positive bacteria.
Levofloxacin has been shown to be active against most isolates of the following bacteria both in vitro and in clinical infections as described in Indications and Usage (1):
Gram-Positive Bacteria
- Enterococcus faecalis
- Staphylococcus aureus (methicillin-susceptible isolates)
- Staphylococcus epidermidis (methicillin-susceptible isolates)
- Staphylococcus saprophyticus
- Streptococcus pneumoniae (including multi-drug resistant isolates [MDRSP]1)
- Streptococcus pyogenes
Gram-Negative Bacteria
- Enterobacter cloacae
- Escherichia coli
- Haemophilus influenzae
- Haemophilus parainfluenzae
- Klebsiella pneumoniae
- Legionella pneumophila
- Moraxella catarrhalis
- Proteus mirabilis
- Pseudomonas aeruginosa
- Serratia marcescens
Other Bacteria
- Chlamydophila pneumoniae
- Mycoplasma pneumoniae
The following in vitro data are available, but their clinical significance is unknown: Levofloxacin exhibits in vitro minimum inhibitory concentrations (MIC values) of 2 mcg/mL or less against most (≥90%) isolates of the following microorganisms; however, the safety and effectiveness of levofloxacin in treating clinical infections due to these bacteria have not been established in adequate and well-controlled clinical trials.
Gram-Positive Bacteria
- Staphylococcus haemolyticus
- β-hemolytic Streptococcus (Group C/F)
- β-hemolytic Streptococcus (Group G)
- Streptococcus agalactiae
- Streptococcus milleri
- Viridans group streptococci
- Bacillus anthracis
Gram-Negative Bacteria
- Acinetobacter baumannii
- Acinetobacter lwoffii
- Bordetella pertussis
- Citrobacter koseri
- Citrobacter freundii
- Enterobacter aerogenes
- Enterobacter sakazakii
- Klebsiella oxytoca
- Morganella morganii
- Pantoea agglomerans
- Proteus vulgaris
- Providencia rettgeri
- Providencia stuartii
- Pseudomonas fluorescens
- Yersinia pestis
Anaerobic Gram-Positive Bacteria
- Clostridium perfringens
- 1
- MDRSP (Multi-drug resistant Streptococcus pneumoniae) isolates are isolates resistant to two or more of the following antibiotics: penicillin (MIC ≥2 mcg/mL), 2nd generation cephalosporins, e.g., cefuroxime; macrolides, tetracyclines and trimethoprim/sulfamethoxazole.
Find Levofloxacin in 5% Dextrose Injection, USP medical information:
Find Levofloxacin in 5% Dextrose Injection, USP medical information:
Levofloxacin in 5% Dextrose Injection, USP Quick Finder
Health Professional Information
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Levofloxacin is a member of the fluoroquinolone class of antibacterial agents [see Microbiology (12.4)].
12.3 Pharmacokinetics
The mean ± SD pharmacokinetic parameters of levofloxacin determined under single and steady-state conditions following oral tablet, oral solution, or intravenous (IV) doses of levofloxacin are summarized in Table 8.
| Regimen | Cmax (mcg/mL) | Tmax (h) | AUC (mcg∙h/mL) | CL/F* (mL/min) | Vd/F† (L) | t1/2 (h) | CLR (mL/min) |
|---|---|---|---|---|---|---|---|
| |||||||
| Single dose | |||||||
| 250 mg oral tablet‡ | 2.8 ± 0.4 | 1.6 ± 1.0 | 27.2 ± 3.9 | 156 ± 20 | ND | 7.3 ± 0.9 | 142 ± 21 |
| 500 mg oral tablet‡§ | 5.1 ± 0.8 | 1.3 ± 0.6 | 47.9 ± 6.8 | 178 ± 28 | ND | 6.3 ± 0.6 | 103 ± 30 |
| 500 mg oral solution¶ | 5.8 ± 1.8 | 0.8 ± 0.7 | 47.8 ± 10.8 | 183 ± 40 | 112 ± 37.2 | 7.0 ± 1.4 | ND |
| 500 mg IV‡ | 6.2 ± 1.0 | 1.0 ± 0.1 | 48.3 ± 5.4 | 175 ± 20 | 90 ± 11 | 6.4 ± 0.7 | 112 ± 25 |
| 750 mg oral tablet#§ | 9.3 ± 1.6 | 1.6 ± 0.8 | 101 ± 20 | 129 ± 24 | 83 ± 17 | 7.5 ± 0.9 | ND |
| 750 mg IV# | 11.5 ± 4.0Þ | ND | 110 ± 40 | 126 ± 39 | 75 ± 13 | 7.5 ± 1.6 | ND |
| Multiple dose | |||||||
| 500 mg every 24h oral tablet‡ | 5.7 ± 1.4 | 1.1 ± 0.4 | 47.5 ± 6.7 | 175 ± 25 | 102 ± 22 | 7.6 ± 1.6 | 116 ± 31 |
| 500 mg every 24h IV‡ | 6.4 ± 0.8 | ND | 54.6 ± 11.1 | 158 ± 29 | 91 ± 12 | 7.0 ± 0.8 | 99 ± 28 |
| 500 mg or 250 mg every 24h IV, patients with bacterial infectionß | 8.7 ± 4.0à | ND | 72.5 ± 51.2à | 154 ± 72 | 111 ± 58 | ND | ND |
| 750 mg every 24h oral tablet# | 8.6 ± 1.9 | 1.4 ± 0.5 | 90.7 ± 17.6 | 143 ± 29 | 100 ± 16 | 8.8 ± 1.5 | 116 ± 28 |
| 750 mg every 24h IV# | 12.1 ± 4.1Þ | ND | 108 ± 34 | 126 ± 37 | 80 ± 27 | 7.9 ± 1.9 | ND |
| 500 mg oral tablet single dose, effects of gender and age: | |||||||
| Maleè | 5.5 ± 1.1 | 1.2 ± 0.4 | 54.4 ± 18.9 | 166 ± 44 | 89 ± 13 | 7.5 ± 2.1 | 126 ± 38 |
| Femaleð | 7.0 ± 1.6 | 1.7 ± 0.5 | 67.7 ± 24.2 | 136 ± 44 | 62 ± 16 | 6.1 ± 0.8 | 106 ± 40 |
| Youngø | 5.5 ± 1.0 | 1.5 ± 0.6 | 47.5 ± 9.8 | 182 ± 35 | 83 ± 18 | 6.0 ± 0.9 | 140 ± 33 |
| Elderlyý | 7.0 ± 1.6 | 1.4 ± 0.5 | 74.7 ± 23.3 | 121 ± 33 | 67 ± 19 | 7.6 ± 2.0 | 91 ± 29 |
| 500 mg oral single dose tablet, patients with renal insufficiency: | |||||||
| CLCR 50–80 mL/min | 7.5 ± 1.8 | 1.5 ± 0.5 | 95.6 ± 11.8 | 88 ± 10 | ND | 9.1 ± 0.9 | 57 ± 8 |
| CLCR 20–49 mL/min | 7.1 ± 3.1 | 2.1 ± 1.3 | 182.1 ± 62.6 | 51 ± 19 | ND | 27 ± 10 | 26 ± 13 |
| CLCR <20 mL/min | 8.2 ± 2.6 | 1.1 ± 1.0 | 263.5 ± 72.5 | 33 ± 8 | ND | 35 ± 5 | 13 ± 3 |
| Hemodialysis | 5.7 ± 1.0 | 2.8 ± 2.2 | ND | ND | ND | 76 ± 42 | ND |
| CAPD | 6.9 ± 2.3 | 1.4 ± 1.1 | ND | ND | ND | 51 ± 24 | ND |
Absorption
Levofloxacin is rapidly and essentially completely absorbed after oral administration. Peak plasma concentrations are usually attained one to two hours after oral dosing. The absolute bioavailability of levofloxacin from a 500 mg tablet and a 750 mg tablet of levofloxacin are both approximately 99%, demonstrating complete oral absorption of levofloxacin. Following a single intravenous dose of levofloxacin to healthy volunteers, the mean ± SD peak plasma concentration attained was 6.2 ± 1.0 mcg/mL after a 500 mg dose infused over 60 minutes and 11.5 ± 4.0 mcg/mL after a 750 mg dose infused over 90 minutes. Levofloxacin Oral Solution and Tablet formulations are bioequivalent.
Levofloxacin pharmacokinetics are linear and predictable after single and multiple oral or IV dosing regimens. Steady-state conditions are reached within 48 hours following a 500 mg or 750 mg once-daily dosage regimen. The mean ± SD peak and trough plasma concentrations attained following multiple once-daily oral dosage regimens were approximately 5.7 ± 1.4 and 0.5 ± 0.2 mcg/mL after the 500 mg doses, and 8.6 ± 1.9 and 1.1 ± 0.4 mcg/mL after the 750 mg doses, respectively. The mean ± SD peak and trough plasma concentrations attained following multiple once-daily IV regimens were approximately 6.4 ± 0.8 and 0.6 ± 0.2 mcg/mL after the 500 mg doses, and 12.1 ± 4.1 and 1.3 ± 0.71 mcg/mL after the 750 mg doses, respectively. Oral administration of a 500 mg dose of levofloxacin with food prolongs the time to peak concentration by approximately 1 hour and decreases the peak concentration by approximately 14% following tablet and approximately 25% following oral solution administration. Therefore, levofloxacin tablets can be administered without regard to food. It is recommended that levofloxacin oral solution be taken 1 hour before or 2 hours after eating.
The plasma concentration profile of levofloxacin after IV administration is similar and comparable in extent of exposure (AUC) to that observed for levofloxacin tablets when equal doses (mg/mg) are administered. Therefore, the oral and IV routes of administration can be considered interchangeable (see Figure 2 and Figure 3).
| Figure 2: Mean Levofloxacin Plasma Concentration vs. Time Profile: 750 mg |
|
| Figure 3: Mean Levofloxacin Plasma Concentration vs. Time Profile: 500 mg |
|
Distribution
The mean volume of distribution of levofloxacin generally ranges from 74 to 112 L after single and multiple 500 mg or 750 mg doses, indicating widespread distribution into body tissues. Levofloxacin reaches its peak levels in skin tissues and in blister fluid of healthy subjects at approximately 3 hours after dosing. The skin tissue biopsy to plasma AUC ratio is approximately 2 and the blister fluid to plasma AUC ratio is approximately 1 following multiple once-daily oral administration of 750 mg and 500 mg doses of levofloxacin, respectively, to healthy subjects. Levofloxacin also penetrates well into lung tissues. Lung tissue concentrations were generally 2- to 5-fold higher than plasma concentrations and ranged from approximately 2.4 to 11.3 mcg/g over a 24-hour period after a single 500 mg oral dose.
In vitro, over a clinically relevant range (1 to 10 mcg/mL) of serum/plasma levofloxacin concentrations, levofloxacin is approximately 24 to 38% bound to serum proteins across all species studied, as determined by the equilibrium dialysis method. Levofloxacin is mainly bound to serum albumin in humans. Levofloxacin binding to serum proteins is independent of the drug concentration.
Metabolism
Levofloxacin is stereochemically stable in plasma and urine and does not invert metabolically to its enantiomer, D-ofloxacin. Levofloxacin undergoes limited metabolism in humans and is primarily excreted as unchanged drug in the urine. Following oral administration, approximately 87% of an administered dose was recovered as unchanged drug in urine within 48 hours, whereas less than 4% of the dose was recovered in feces in 72 hours. Less than 5% of an administered dose was recovered in the urine as the desmethyl and N-oxide metabolites, the only metabolites identified in humans. These metabolites have little relevant pharmacological activity.
Excretion
Levofloxacin is excreted largely as unchanged drug in the urine. The mean terminal plasma elimination half-life of levofloxacin ranges from approximately 6 to 8 hours following single or multiple doses of levofloxacin given orally or intravenously. The mean apparent total body clearance and renal clearance range from approximately 144 to 226 mL/min and 96 to 142 mL/min, respectively. Renal clearance in excess of the glomerular filtration rate suggests that tubular secretion of levofloxacin occurs in addition to its glomerular filtration. Concomitant administration of either cimetidine or probenecid results in approximately 24% and 35% reduction in the levofloxacin renal clearance, respectively, indicating that secretion of levofloxacin occurs in the renal proximal tubule. No levofloxacin crystals were found in any of the urine samples freshly collected from subjects receiving levofloxacin.
Geriatric
There are no significant differences in levofloxacin pharmacokinetics between young and elderly subjects when the subjects' differences in creatinine clearance are taken into consideration. Following a 500 mg oral dose of levofloxacin to healthy elderly subjects (66 – 80 years of age), the mean terminal plasma elimination half-life of levofloxacin was about 7.6 hours, as compared to approximately 6 hours in younger adults. The difference was attributable to the variation in renal function status of the subjects and was not believed to be clinically significant. Drug absorption appears to be unaffected by age. Levofloxacin dose adjustment based on age alone is not necessary [see Use in Specific Populations (8.5)].
Pediatrics
The pharmacokinetics of levofloxacin following a single 7 mg/kg intravenous dose were investigated in pediatric patients ranging in age from 6 months to 16 years. Pediatric patients cleared levofloxacin faster than adult patients, resulting in lower plasma exposures than adults for a given mg/kg dose. Subsequent pharmacokinetic analyses predicted that a dosage regimen of 8 mg/kg every 12 hours (not to exceed 250 mg per dose) for pediatric patients 6 months to 17 years of age would achieve comparable steady state plasma exposures (AUC0–24 and Cmax) to those observed in adult patients administered 500 mg of levofloxacin once every 24 hours.
Gender
There are no significant differences in levofloxacin pharmacokinetics between male and female subjects when subjects' differences in creatinine clearance are taken into consideration. Following a 500 mg oral dose of levofloxacin to healthy male subjects, the mean terminal plasma elimination half-life of levofloxacin was about 7.5 hours, as compared to approximately 6.1 hours in female subjects. This difference was attributable to the variation in renal function status of the male and female subjects and was not believed to be clinically significant. Drug absorption appears to be unaffected by the gender of the subjects. Dose adjustment based on gender alone is not necessary.
Race
The effect of race on levofloxacin pharmacokinetics was examined through a covariate analysis performed on data from 72 subjects: 48 white and 24 non-white. The apparent total body clearance and apparent volume of distribution were not affected by the race of the subjects.
Renal Impairment
Clearance of levofloxacin is substantially reduced and plasma elimination half-life is substantially prolonged in adult patients with impaired renal function (creatinine clearance <50 mL/min), requiring dosage adjustment in such patients to avoid accumulation. Neither hemodialysis nor continuous ambulatory peritoneal dialysis (CAPD) is effective in removal of levofloxacin from the body, indicating that supplemental doses of levofloxacin are not required following hemodialysis or CAPD [see Dosage and Administration (2.3), Use in Specific Populations (8.6)].
Hepatic Impairment
Pharmacokinetic studies in hepatically impaired patients have not been conducted. Due to the limited extent of levofloxacin metabolism, the pharmacokinetics of levofloxacin are not expected to be affected by hepatic impairment [see Use in Specific Populations (8.7)].
Bacterial Infection
The pharmacokinetics of levofloxacin in patients with serious community-acquired bacterial infections are comparable to those observed in healthy subjects.
Drug-Drug Interactions
The potential for pharmacokinetic drug interactions between levofloxacin and antacids, warfarin, theophylline, cyclosporine, digoxin, probenecid, and cimetidine has been evaluated [see Drug Interactions (7)].
12.4 Microbiology
Mechanism of Action
Levofloxacin is the L-isomer of the racemate, ofloxacin, a quinolone antimicrobial agent. The antibacterial activity of ofloxacin resides primarily in the L-isomer. The mechanism of action of levofloxacin and other fluoroquinolone antimicrobials involves inhibition of bacterial topoisomerase IV and DNA gyrase (both of which are type II topoisomerases), enzymes required for DNA replication, transcription, repair and recombination.
Resistance
Fluoroquinolone resistance can arise through mutations in defined regions of DNA gyrase or topoisomerase IV, termed the Quinolone-Resistance Determining Regions (QRDRs), or through altered efflux.
Fluoroquinolones, including levofloxacin, differ in chemical structure and mode of action from aminoglycosides, macrolides and β-lactam antibiotics, including penicillins. Fluoroquinolones may, therefore, be active against bacteria resistant to these antimicrobials.
Resistance to levofloxacin due to spontaneous mutation in vitro is a rare occurrence (range: 10-9 to 10-10). Cross-resistance has been observed between levofloxacin and some other fluoroquinolones, some microorganisms resistant to other fluoroquinolones may be susceptible to levofloxacin.
Antimicrobial Activity
Levofloxacin has in vitro activity against Gram-negative and Gram-positive bacteria.
Levofloxacin has been shown to be active against most isolates of the following bacteria both in vitro and in clinical infections as described in Indications and Usage (1):
Gram-Positive Bacteria
- Enterococcus faecalis
- Staphylococcus aureus (methicillin-susceptible isolates)
- Staphylococcus epidermidis (methicillin-susceptible isolates)
- Staphylococcus saprophyticus
- Streptococcus pneumoniae (including multi-drug resistant isolates [MDRSP]1)
- Streptococcus pyogenes
Gram-Negative Bacteria
- Enterobacter cloacae
- Escherichia coli
- Haemophilus influenzae
- Haemophilus parainfluenzae
- Klebsiella pneumoniae
- Legionella pneumophila
- Moraxella catarrhalis
- Proteus mirabilis
- Pseudomonas aeruginosa
- Serratia marcescens
Other Bacteria
- Chlamydophila pneumoniae
- Mycoplasma pneumoniae
The following in vitro data are available, but their clinical significance is unknown: Levofloxacin exhibits in vitro minimum inhibitory concentrations (MIC values) of 2 mcg/mL or less against most (≥90%) isolates of the following microorganisms; however, the safety and effectiveness of levofloxacin in treating clinical infections due to these bacteria have not been established in adequate and well-controlled clinical trials.
Gram-Positive Bacteria
- Staphylococcus haemolyticus
- β-hemolytic Streptococcus (Group C/F)
- β-hemolytic Streptococcus (Group G)
- Streptococcus agalactiae
- Streptococcus milleri
- Viridans group streptococci
- Bacillus anthracis
Gram-Negative Bacteria
- Acinetobacter baumannii
- Acinetobacter lwoffii
- Bordetella pertussis
- Citrobacter koseri
- Citrobacter freundii
- Enterobacter aerogenes
- Enterobacter sakazakii
- Klebsiella oxytoca
- Morganella morganii
- Pantoea agglomerans
- Proteus vulgaris
- Providencia rettgeri
- Providencia stuartii
- Pseudomonas fluorescens
- Yersinia pestis
Anaerobic Gram-Positive Bacteria
- Clostridium perfringens
- 1
- MDRSP (Multi-drug resistant Streptococcus pneumoniae) isolates are isolates resistant to two or more of the following antibiotics: penicillin (MIC ≥2 mcg/mL), 2nd generation cephalosporins, e.g., cefuroxime; macrolides, tetracyclines and trimethoprim/sulfamethoxazole.
Health Professional Information
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5Pm ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.