EUCRISA
(crisaborole)
Find EUCRISA medical information:
Find EUCRISA medical information:
EUCRISA Quick Finder
Highlights
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use EUCRISA safely and effectively. See full prescribing information for EUCRISA. EUCRISA (crisaborole) ointment, for topical use Initial U.S. Approval: 2016 INDICATIONS AND USAGEEUCRISA is a phosphodiesterase 4 inhibitor indicated for topical treatment of mild to moderate atopic dermatitis in adult and pediatric patients 3 months of age and older. (1) DOSAGE AND ADMINISTRATIONDOSAGE FORMS AND STRENGTHSOintment, 2%. (3) CONTRAINDICATIONSKnown hypersensitivity to crisaborole or any component of the formulation. (4) WARNINGS AND PRECAUTIONSHypersensitivity reactions: If signs and symptoms of hypersensitivity occur, discontinue EUCRISA immediately and initiate appropriate therapy. (5.1) ADVERSE REACTIONSThe most common adverse reaction occurring in ≥1% in subjects is application site pain. (6.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 4/2023 |
Indications and Usage
Dosage and Administration
2 DOSAGE AND ADMINISTRATION
Apply a thin layer of EUCRISA twice daily to affected areas. Once clinical effect is achieved, consider reducing application to once daily [see Clinical Studies (14)].
EUCRISA is for topical use only and not for ophthalmic, oral, or intravaginal use.
Dosage Forms and Strengths
Contraindications
4 CONTRAINDICATIONS
EUCRISA is contraindicated in patients with known hypersensitivity to crisaborole or any component of the formulation. [see Warnings and Precautions (5.1)]
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions, including contact urticaria, have occurred in patients treated with EUCRISA. Hypersensitivity should be suspected in the event of severe pruritus, swelling and erythema at the application site or at a distant site. If signs and symptoms of hypersensitivity occur, discontinue EUCRISA immediately and initiate appropriate therapy.
Adverse Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In two double-blind, vehicle-controlled clinical trials (Trial 1 and Trial 2), 1012 subjects 2 to 79 years of age with mild to moderate atopic dermatitis were treated with EUCRISA twice daily for 4 weeks. The adverse reaction reported by ≥1% of EUCRISA-treated subjects is listed in Table 1.
| Adverse Reaction | EUCRISA Twice Daily N=1012 n (%) | Vehicle Twice Daily N=499 n (%) |
|---|---|---|
| ||
Application site pain* | 45 (4) | 6 (1) |
Less common (<1%) adverse reactions in subjects treated with EUCRISA included contact urticaria [see Warnings and Precautions (5.1)].
In one double-blind, vehicle-controlled trial including an initial open-label period (Trial 3), 497 subjects 3 months of age and older with mild to moderate atopic dermatitis received EUCRISA twice daily for up to 8 weeks. This was followed by a double-blind period, during which 135 subjects out of 270 randomized subjects received EUCRISA and 135 subjects received vehicle once daily for 52 weeks or until they developed a flare. The adverse reactions observed in the open-label period were similar to the known safety profile of twice daily treatment with EUCRISA. The adverse reactions observed with once daily treatment were similar to vehicle [see Clinical Studies (14)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of EUCRISA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
Skin and Subcutaneous: allergic contact dermatitis
Use in Specific Populations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from case reports with EUCRISA use in pregnant women are insufficient to inform a drug-associated risk for major birth defects, miscarriage, or other adverse maternal or fetal outcomes. In animal reproduction studies, there were no adverse developmental effects observed with oral administration of crisaborole in pregnant rats and rabbits during organogenesis at doses up to 3 and 2 times, respectively, the maximum recommended human dose (MRHD) (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies carry some risk of birth defect, loss, or other adverse outcomes. The background risk of major birth defects in the U.S. general population is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Animal Data
Rat and rabbit embryo-fetal development was assessed after oral administration of crisaborole. Crisaborole did not cause adverse effects to the fetus at oral doses up to 300 mg/kg/day in pregnant rats during the period of organogenesis (3 times the MRHD on an area under the curve (AUC) comparison basis). No crisaborole-related fetal malformations were noted after oral treatment with crisaborole in pregnant rats at doses up to 600 mg/kg/day (13 times the MRHD on an AUC comparison basis) during the period of organogenesis. Maternal toxicity was produced at this high dose of 600 mg/kg/day in pregnant rats and was associated with decreased fetal body weight and delayed skeletal ossification. Crisaborole did not cause adverse effects to the fetus at oral doses up to the highest dose tested of 100 mg/kg/day in pregnant rabbits during the period of organogenesis (2 times the MRHD on an AUC comparison basis).
In a prenatal/postnatal development study, pregnant rats were treated with crisaborole at doses of 150, 300, or 600 mg/kg/day by oral gavage during gestation and lactation (from gestation day 7 through day 20 of lactation). Crisaborole did not have any adverse effects on fetal development at doses up to 300 mg/kg/day (3 times the MRHD on an AUC comparison basis). Maternal toxicity was produced at the high dose of 600 mg/kg/day in pregnant rats and was associated with stillbirths, pup mortality, and reduced pup weights.
8.2 Lactation
Risk Summary
There is no information available on the presence of EUCRISA in human milk, the effects of the drug on the breastfed infant or the effects of the drug on milk production after topical application of EUCRISA to women who are breastfeeding. EUCRISA is systemically absorbed. The lack of clinical data during lactation precludes a clear determination of the risk of EUCRISA to a breastfed infant. Therefore, the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for EUCRISA and any potential adverse effects on the breastfed infant from EUCRISA or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of EUCRISA have been established in pediatric patients ages 3 months and older for topical treatment of mild to moderate atopic dermatitis. Use of EUCRISA administered twice daily in this age group is supported by data from two 28-day adequate, vehicle-controlled safety and efficacy trials (1,313 pediatric subjects ages 2 years to 17 years of whom 874 received EUCRISA), a 28-day open-label, safety and pharmacokinetics (PK) trial (137 subjects ages 3 months to less than 2 years who received EUCRISA), and another trial with an open-label period of up to 8 weeks (327 pediatric subjects ages 5 months to less than 18 years who received EUCRISA) [see Clinical Pharmacology (12.3) and Clinical Studies (14)].
The safety and effectiveness of EUCRISA in pediatric patients below the age of 3 months have not been established.
Description
11 DESCRIPTION
EUCRISA contains 2% crisaborole (w/w) in a petrolatum-based, white to off-white ointment and is for topical use. The active ingredient, crisaborole, is a phosphodiesterase-4 (PDE-4) inhibitor.
Crisaborole is described chemically as 5-(4-cyanophenoxy)-1,3-dihydro-1-hydroxy-[2,1]-benzoxaborole. The empirical formula is C14H10BNO3 and the molecular weight is 251.1 g/mol.
The structural formula is represented below:
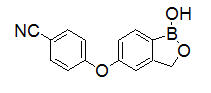
Crisaborole drug substance is freely soluble in common organic solvents such as isopropyl alcohol and propylene glycol, and insoluble in water.
Each gram of EUCRISA contains 20 mg of crisaborole in an ointment containing white petrolatum, propylene glycol, mono- and di-glycerides, paraffin, butylated hydroxytoluene, and edetate calcium disodium.
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Crisaborole is a phosphodiesterase 4 (PDE-4) inhibitor. PDE-4 inhibition results in increased intracellular cyclic adenosine monophosphate (cAMP) levels. The specific mechanism(s) by which crisaborole exerts its therapeutic action for the treatment of atopic dermatitis is not well defined.
12.3 Pharmacokinetics
Absorption
The PK of EUCRISA were investigated in 33 pediatric subjects 2 to 17 years of age with mild to moderate atopic dermatitis and a mean ± SD body surface area (BSA) involvement of 49 ± 20% (range 27% to 92%). In this study, subjects applied approximately 3 mg/cm2 of EUCRISA ointment (dose range was approximately 6 g to 30 g per application) twice daily for 8 days.
Plasma concentrations were quantifiable in all the subjects. The mean ± SD maximum plasma concentration (Cmax) and area under the concentration time curve from 0 to 12 hours post dose (AUC0–12) for crisaborole on Day 8 were 127 ± 196 ng/mL and 949 ± 1240 ng h/mL, respectively. Systemic concentrations of crisaborole were at steady state by Day 8. Based on the ratios of AUC0–12 between Day 8 and Day 1, the mean accumulation factor for crisaborole was 1.9.
The PK of EUCRISA were investigated in 13 subjects 4 months to less than 24 months of age. The mean ± SD Cmax and AUC0–12 for crisaborole were 188 ± 100 ng/mL and 1164 ± 550 ng∙h/mL, respectively.
Elimination
Metabolism
Crisaborole is substantially metabolized into inactive metabolites. The major metabolite 5-(4-cyanophenoxy)-2-hydroxyl benzylalcohol (metabolite 1), is formed via hydrolysis; this metabolite is further metabolized into downstream metabolites, among which 5-(4-cyanophenoxy)-2-hydroxyl benzoic acid (metabolite 2), formed via oxidation, is also a major metabolite.
PK of metabolites 1 and 2 were assessed in the PK study described above and the systemic concentrations were at or near steady state by Day 8. Based on the ratios of AUC0–12 between Day 8 and Day 1, the mean accumulation factors for metabolites 1 and 2 were 1.7 and 6.3, respectively.
Drug Interaction Studies
In vitro studies using human liver microsomes indicated that under the conditions of clinical use, crisaborole and metabolite 1 are not expected to inhibit cytochrome P450 (CYP) 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4.
In vitro human liver microsomes studies for metabolite 2 showed that it did not inhibit activities of CYP2C19, 2D6, and 3A4; was a weak inhibitor of CYP1A2 and 2B6; and a moderate inhibitor of CYP2C8 and 2C9. The most sensitive enzyme, CYP2C9, was further investigated in a clinical trial using warfarin as a CYP2C9 substrate. The results of this study showed no drug interaction potential.
In vitro studies in human hepatocytes showed that under the conditions of clinical use, crisaborole and metabolites 1 and 2 are not expected to induce CYP enzymes.
In vitro studies showed that crisaborole and metabolite 1 did not inhibit the activities of uridine diphosphate (UDP)-glucuronosyltransferase (UGT) 1A1, 1A4, 1A6, 1A9, 2B7, and 2B15. Metabolite 2 did not inhibit UGT1A4, 1A6, 2B7, and 2B15. Metabolite 2 showed weak inhibition of UGT1A1, however, no clinically significant drug interactions are expected between crisaborole (and its metabolites) and UGT1A1 substrates at therapeutic concentrations. Metabolite 2 showed moderate inhibition of UGT1A9 and may result in a moderate increase of the concentrations of sensitive UGT1A9 substrates.
In vitro studies indicate that under the condition of clinical use, crisaborole and metabolites 1 and 2 are not expected to cause clinically significant interactions with substrates of P-glycoprotein and organic anionic or cationic transporters. Crisaborole and metabolite 1 are not expected to inhibit breast cancer resistance protein (BCRP); metabolite 2 is expected to inhibit BCRP at therapeutic concentrations.
Nonclinical Toxicology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In an oral carcinogenicity study in Sprague-Dawley rats, oral doses of 30, 100, or 300 mg/kg/day crisaborole were administered to rats once daily. A crisaborole-related increased incidence of benign granular cell tumors in the uterus with cervix and vagina (combined) was noted in 300 mg/kg/day crisaborole treated female rats (2 times the MRHD on an AUC comparison basis). The clinical relevance of this finding is unknown.
In a dermal carcinogenicity study in CD-1 mice, topical doses of 2%, 5%, or 7% crisaborole ointment were administered once daily. No crisaborole-related neoplastic findings were noted at topical doses up to 7% crisaborole ointment (1 times the MRHD on an AUC comparison basis).
Crisaborole revealed no evidence of mutagenic or clastogenic potential based on the results of two in vitro genotoxicity tests (Ames assay and human lymphocyte chromosomal aberration assay) and one in vivo genotoxicity test (rat micronucleus assay).
No effects on fertility were observed in male or female rats that were administered oral doses up to 600 mg/kg/day crisaborole (13 times the MRHD on an AUC comparison basis) prior to and during early pregnancy.
Clinical Studies
14 CLINICAL STUDIES
Two multicenter, randomized, double-blind, parallel-group, vehicle-controlled trials (Trials 1 and 2) treated a total of 1522 subjects 2 to 79 years of age (86.3% of subjects were 2 to 17 years of age) with a 5% to 95% treatable BSA. At baseline, 38.5% of the subjects had an Investigator's Static Global Assessment [ISGA] of mild (2), and 61.5% had an ISGA of moderate (3), in the overall assessment of atopic dermatitis (erythema, induration/papulation, and oozing/crusting) on a severity scale of 0 to 4.
In both trials, subjects were randomized 2:1 to receive EUCRISA or vehicle applied twice daily for 28 days. The primary efficacy endpoint was the proportion of subjects at Day 29 who achieved success, defined as an ISGA grade of clear (0) or almost clear (1) with a 2-grade or greater improvement from baseline, comparing EUCRISA-treated subjects to vehicle-treated subjects.
Efficacy results from the two trials are summarized in Table 2.
| Trial 1 | Trial 2 | |||
|---|---|---|---|---|
| EUCRISA Twice Daily (N=503) | Vehicle Twice Daily (N=256) | EUCRISA Twice Daily (N=513) | Vehicle Twice Daily (N=250) | |
| ||||
Success in ISGA* | 32.8% | 25.4% | 31.4% | 18.0% |
The success rates over time are presented in Figure 1.
| |
Figure 1: Success in ISGA* Over Time in Subjects with Mild to Moderate Atopic Dermatitis | |
Trial 1 | Trial 2 |
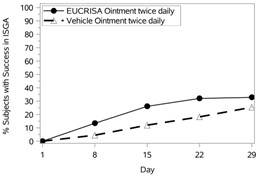 | 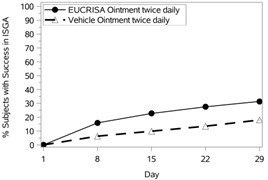 |
One randomized, double-blind, vehicle-controlled trial (Trial 3) assessed the efficacy and safety of EUCRISA once daily over 52 weeks in pediatric (3 months to less than 18 years of age) and adult subjects with mild to moderate atopic dermatitis, who achieved success on EUCRISA twice daily during open-label treatment of up to 8 weeks.
A total of 497 subjects 3 months of age and older with a 2% to 90% treatable BSA, entered into an open-label period to receive EUCRISA twice daily for up to 8 weeks. At baseline, 327 (66%) of subjects were 3 months to less than 18 years of age, 66% of the subjects had an ISGA of moderate (3), and 34% had an ISGA of mild (2), in the overall assessment of atopic dermatitis (erythema, induration/papulation, and oozing/crusting) on a severity scale of 0 to 4.
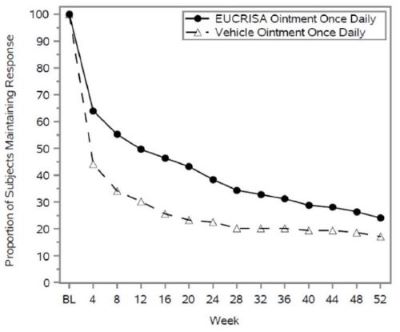
Of the 497, a total of 254 subjects 3 months of age and older, who achieved both ISGA success (score of clear [0] or almost clear [1] with a ≥2 grade improvement from baseline) and EASI50 response (at least 50% improvement from baseline in EASI scores) were randomized 1:1 into a double-blind period to receive EUCRISA once daily or vehicle for 52 weeks or until they developed a flare. At the beginning of the double-blind period, 59% of the subjects had an ISGA of almost clear (1) and 41% had an ISGA of clear (0).
Figure 2 presents the percentage of subjects maintaining an ISGA of clear or almost clear through Week 52.
Figure 2: Percentage of Subjects Maintaining ISGA of Clear or Almost Clear Through Week 52
How Supplied/Storage and Handling
Medication Guide
17 PATIENT COUNSELING INFORMATION
Advise the patient or caregivers to read the FDA-approved patient labeling (Patient Information).
Hypersensitivity Reactions
Advise patients to discontinue EUCRISA and seek medical attention immediately if signs or symptoms of hypersensitivity occur [see Warnings and Precautions (5.1)].
This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 4/2023 | |
PATIENT INFORMATION | ||
Important information: EUCRISA is for use on skin (topical use) only. Do not use EUCRISA in your eyes, mouth, or vagina. | ||
What is EUCRISA? EUCRISA is a prescription medicine used on the skin (topical) to treat mild to moderate eczema (atopic dermatitis) in adults and children 3 months of age and older. It is not known if EUCRISA is safe and effective in children under 3 months of age. | ||
Who should not use EUCRISA? Do not use EUCRISA if you are allergic to crisaborole or any of the ingredients in EUCRISA. See the end of this Patient Information leaflet for a complete list of ingredients in EUCRISA. | ||
Before using EUCRISA, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. | ||
How should I use EUCRISA?
| ||
What are the possible side effects of EUCRISA? EUCRISA may cause side effects.
| ||
|
| |
|
| |
|
| |
The most common side effect of EUCRISA is application site pain, such as burning or stinging. This is not the only possible side effect of EUCRISA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | ||
How should I store EUCRISA?
Keep EUCRISA and all medicines out of the reach of children. | ||
General information about the safe and effective use of EUCRISA Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use EUCRISA for a condition for which it was not prescribed. Do not give EUCRISA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about EUCRISA that is written for healthcare professionals. | ||
What are the ingredients in EUCRISA? Active ingredient: crisaborole Inactive ingredients: white petrolatum, propylene glycol, mono- and di-glycerides, paraffin, butylated hydroxytoluene, and edetate calcium disodium.  LAB-0917-5.0 For more information, call 1-866-EUCRISA [1-866-382-7472] or go to www.EUCRISA.com | ||
Other

What is EUCRISA?
What is EUCRISA?
EUCRISA is a prescription medicine used on the skin (topical) to treat mild to moderate eczema (atopic dermatitis) in adults and children 3 months of age and older.
It is not known if EUCRISA is safe and effective in children under 3 months of age.
Who should not use EUCRISA?
Who should not use EUCRISA?
Do not use EUCRISA if you are allergic to crisaborole or any of the ingredients in EUCRISA. See the end of this Patient Information leaflet for a complete list of ingredients in EUCRISA.
Before using EUCRISA, tell your healthcare provider about all of your medical conditions, including if you:
- •
- are pregnant or plan to become pregnant. It is not known if EUCRISA will harm your unborn baby.
- •
- are breastfeeding or plan to breastfeed. It is not known if EUCRISA passes into your breast milk.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How should I use EUCRISA?
How should I use EUCRISA?
- •
- Use EUCRISA exactly as your healthcare provider tells you to use it.
- •
- Apply a thin layer of EUCRISA to the affected areas 2 times each day or as directed by your healthcare provider.
- •
- Wash your hands after applying EUCRISA, unless hands are being treated. If someone else applies EUCRISA for you, they should wash their hands after applying EUCRISA.
What are the possible side effects of EUCRISA?
What are the possible side effects of EUCRISA?
EUCRISA may cause side effects.
- •
- Allergic reactions. EUCRISA may cause allergic reactions at or near the application site or at a distant site which may be serious. Stop using EUCRISA and get medical help right away if you have any symptoms of an allergic reaction including:
- o
- trouble breathing or throat tightness
- o
- hives
- o
- chest tightness
- o
- itching
- o
- feeling faint
- o
- swelling of your face, eyelids, lips, mouth, tongue or throat
- o
- redness
The most common side effect of EUCRISA is application site pain, such as burning or stinging.
This is not the only possible side effect of EUCRISA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store EUCRISA?
How should I store EUCRISA?
- •
- Store EUCRISA at room temperature, between 68°F to 77°F (20°C to 25°C).
- •
- Keep the tube tightly closed.
Keep EUCRISA and all medicines out of the reach of children.
General information about the safe and effective use of EUCRISA
General information about the safe and effective use of EUCRISA
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use EUCRISA for a condition for which it was not prescribed. Do not give EUCRISA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about EUCRISA that is written for healthcare professionals.
What are the ingredients in EUCRISA?
What are the ingredients in EUCRISA?
Active ingredient: crisaborole
Inactive ingredients: white petrolatum, propylene glycol, mono- and di-glycerides, paraffin, butylated hydroxytoluene, and edetate calcium disodium.
LAB-0917-5.0
For more information, call 1-866-EUCRISA [1-866-382-7472] or go to www.EUCRISA.com
Full Patient Information
Full Patient Information
17 PATIENT COUNSELING INFORMATION
Advise the patient or caregivers to read the FDA-approved patient labeling (Patient Information).
Hypersensitivity Reactions
Advise patients to discontinue EUCRISA and seek medical attention immediately if signs or symptoms of hypersensitivity occur [see Warnings and Precautions (5.1)].
This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 4/2023 | |
PATIENT INFORMATION | ||
Important information: EUCRISA is for use on skin (topical use) only. Do not use EUCRISA in your eyes, mouth, or vagina. | ||
What is EUCRISA? EUCRISA is a prescription medicine used on the skin (topical) to treat mild to moderate eczema (atopic dermatitis) in adults and children 3 months of age and older. It is not known if EUCRISA is safe and effective in children under 3 months of age. | ||
Who should not use EUCRISA? Do not use EUCRISA if you are allergic to crisaborole or any of the ingredients in EUCRISA. See the end of this Patient Information leaflet for a complete list of ingredients in EUCRISA. | ||
Before using EUCRISA, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. | ||
How should I use EUCRISA?
| ||
What are the possible side effects of EUCRISA? EUCRISA may cause side effects.
| ||
|
| |
|
| |
|
| |
The most common side effect of EUCRISA is application site pain, such as burning or stinging. This is not the only possible side effect of EUCRISA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | ||
How should I store EUCRISA?
Keep EUCRISA and all medicines out of the reach of children. | ||
General information about the safe and effective use of EUCRISA Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use EUCRISA for a condition for which it was not prescribed. Do not give EUCRISA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about EUCRISA that is written for healthcare professionals. | ||
What are the ingredients in EUCRISA? Active ingredient: crisaborole Inactive ingredients: white petrolatum, propylene glycol, mono- and di-glycerides, paraffin, butylated hydroxytoluene, and edetate calcium disodium. LAB-0917-5.0 For more information, call 1-866-EUCRISA [1-866-382-7472] or go to www.EUCRISA.com | ||
Resources
Didn’t find what you were looking for?
Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5Pm ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this product, click the link below to submit your information: Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer product, please call Pfizer Medical Information at (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800)-332-1088.