ERAXIS®
(anidulafungin)
Find ERAXIS® medical information:
Find ERAXIS® medical information:
ERAXIS® Quick Finder
Highlights
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use ERAXIS safely and effectively. See full prescribing information for ERAXIS. ERAXIS (anidulafungin) for injection, for intravenous use Initial U.S. Approval: 2006 INDICATIONS AND USAGEERAXIS is an echinocandin antifungal indicated for the treatment of the following infections:
Limitations of use
DOSAGE AND ADMINISTRATION
Rate of Infusion for Adults and Pediatric Patients The rate of infusion should not exceed 1.1 mg/minute [equivalent to 1.4 mL/minute or 84 mL/hour when reconstituted and diluted per instructions] (2.3, 2.4) DOSAGE FORMS AND STRENGTHSFor injection: 50 mg, and 100 mg as a lyophilized powder in a single-dose vial for reconstitution (3) CONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSAdults
Pediatric Patients (1 month and older) Candidemia and other forms of Candida infections: Most common adverse reactions (≥ 5%): diarrhea, vomiting, pyrexia, abdominal pain, anemia, thrombocytopenia, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) increased, hypoglycemia, epistaxis, and rash. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. USE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 5/2023 | ||||||||||
Indications and Usage
1 INDICATIONS AND USAGE
1.1 Candidemia and Other Forms of Candida Infections (Intra-abdominal Abscess and Peritonitis)
ERAXIS is indicated for the treatment of candidemia and the following Candida infections: intra-abdominal abscess and peritonitis in adults and pediatric patients 1 month of age and older [see Clinical Studies (14.1) and Microbiology (12.4)].
1.2 Esophageal Candidiasis
ERAXIS is indicated for the treatment of esophageal candidiasis in adults [see Indications and Usage (1.3), Clinical Studies (14.2)].
1.3 Limitations of Use
- •
- ERAXIS has not been studied in adult and pediatric patients with endocarditis, osteomyelitis, and meningitis due to Candida, and has not been studied in sufficient numbers of neutropenic patients to determine efficacy in this group. The dosage of ERAXIS for the treatment of Candida dissemination into the CNS and the eye has not been established [see Warning and Precautions (5.3), Use in Specific Populations (8.4)].
- •
- ERAXIS is associated with high relapse rates in esophageal candidiasis [see Clinical Studies (14.2)].
1.4 Usage
Specimens for fungal culture and other relevant laboratory studies (including histopathology) should be obtained prior to therapy to isolate and identify causative organism(s). Therapy may be instituted before the results of the cultures and other laboratory studies are known. However, once these results become available, antifungal therapy should be adjusted accordingly.
Dosage and Administration
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Adults
Candidemia and other Candida infections (intra-abdominal abscess and peritonitis)
The recommended dose is a single 200 mg loading dose of ERAXIS on Day 1, followed by a 100 mg once daily maintenance dose thereafter. Duration of treatment should be based on the patient's clinical response. In general, antifungal therapy should continue for at least 14 days after the last positive culture.
Esophageal Candidiasis
The recommended dose is a single 100 mg loading dose of ERAXIS on Day 1, followed by a 50 mg once daily maintenance dose thereafter. Patients should be treated for a minimum of 14 days and for at least 7 days following resolution of symptoms. Duration of treatment should be based on the patient's clinical response. Because of the risk of relapse of esophageal candidiasis in patients with HIV infection, suppressive antifungal therapy may be considered after a course of treatment.
2.2 Recommended Dosage in Pediatric Patients (1 Month of Age and Older)
Candidemia and other Candida infections (intra-abdominal abscess and peritonitis)
The recommended dose is a single loading dose of 3 mg/kg (not to exceed 200 mg) of ERAXIS on Day 1, followed by a once daily maintenance dose of 1.5 mg/kg (not to exceed 100 mg) of ERAXIS thereafter. Overall antifungal treatment should continue for at least 14 days after the last positive culture.
2.3 Preparation for Administration
ERAXIS for Injection must be reconstituted with sterile Water for Injection and subsequently diluted only with 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP (normal saline), prior to administration.
The compatibility of reconstituted ERAXIS with intravenous substances, additives, or medications other than 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP (normal saline) has not been established. Do NOT dilute with other solutions or co-infuse with other medications or electrolytes. The infusion solution must not be frozen.
Reconstitution of the 50 mg/vial
Aseptically reconstitute each 50 mg vial with 15 mL of sterile Water for Injection to provide a concentration of 3.33 mg/mL.
Reconstitution of the 100 mg/vial
Aseptically reconstitute each 100 mg vial with 30 mL of sterile Water for Injection to provide a concentration of 3.33 mg/mL.
Storage of the Reconstituted Solution
ERAXIS reconstituted solution can be stored at 25°C (77°F) for up to 24 hours prior to dilution into the infusion solution. Chemical and physical in-use stability of the reconstituted solution has been demonstrated for 24 hours at 25°C (77°F). From a microbiological point of view, following good aseptic practices, the reconstituted solution can be utilized for up to 24 hours when stored at 25°C.
2.4 Dilution and Administration of the Infusion
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. If particulate matter or discoloration is identified, discard the solution.
Adult Patients
Aseptically transfer the contents of the reconstituted vial(s) into the appropriately sized IV bag (or bottle) containing either 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP (normal saline). See Table 1 for the dilution to a concentration of 0.77 mg/mL for the final infusion solution and infusion instructions for each dose. The rate of infusion should not exceed 1.1 mg/minute (equivalent to 1.4 mL/minute or 84 mL/hour when reconstituted and diluted per instructions) [see Warnings and Precautions (5.2)].
| Dose | Number of Vials Required | Total Reconstituted Volume Required | Infusion Volume* | Total Infusion Volume† | Rate of Infusion | Minimum Duration of Infusion |
|---|---|---|---|---|---|---|
50 mg | 1–50 mg | 15 mL | 50 mL | 65 mL | 1.4 mL/min or 84 mL/ hour | 45 min |
100 mg | 2–50 mg or 1–100 mg | 30 mL | 100 mL | 130 mL | 1.4 mL/min or 84 mL/ hour | 90 min |
200 mg | 4–50 mg or 2–100 mg | 60 mL | 200 mL | 260 mL | 1.4 mL/min or 84 mL/ hour | 180 min |
Pediatric Patients
The volume of infusion solution required to deliver the dose is dependent on the weight of the child. The reconstituted solution must be further diluted to a concentration of 0.77 mg/mL for the final infusion solution. A programmable syringe or infusion pump is recommended. The rate of infusion should not exceed 1.1 mg/minute (equivalent to 1.4 mL/minute or 84 mL/hour when reconstituted and diluted per instructions) [see Warnings and Precautions (5.2)].
Steps for the Preparation of Pediatric Doses below 50 mg
- 1.
- Calculate pediatric patient dose and aseptically reconstitute vial(s) required according to reconstitution instructions to provide a concentration of 3.33 mg/mL (if dose is 50 mg or above, see preparation instructions for Adult Patients above) [see Dosage and Administration (2.2, 2.3)].
- 2.
- Calculate the volume (mL) of reconstituted ERAXIS required:
[Volume of reconstituted ERAXIS (mL) = Dose of ERAXIS (mg) ÷ 3.33 mg/mL] - 3.
- Calculate the total volume of the infusion solution (mL) that contains a final concentration of 0.77 mg/mL:
[Total volume of infusion solution (mL) = Dose of ERAXIS (mg) ÷ 0.77 mg/mL] - 4.
- Calculate the volume of diluent [5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP (normal saline)] required to prepare the infusion solution: [Volume of diluent (mL) = Total volume of final infusion solution (mL) – Volume of reconstituted ERAXIS (mL)]
- 5.
- Prepare the infusion solution by aseptically transferring the required volumes (mL) of the reconstituted ERAXIS and diluent [5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP (normal saline)] into an infusion syringe or IV infusion bag needed for administration.
Storage of the Infusion Solution
ERAXIS infusion solution can be stored at temperatures up to 25°C (77°F) for up to 48 hours. Do not freeze. Chemical and physical in-use stability of the infusion solution has been demonstrated for 48 hours at 25°C (77°F). From a microbiological point of view, following good aseptic practices, the infusion solution can be utilized for up to 48 hours from preparation when stored at 25°C.
Dosage Forms and Strengths
Contraindications
4 CONTRAINDICATIONS
ERAXIS is contraindicated in:
- •
- Patients with known hypersensitivity to anidulafungin, any component of ERAXIS, or other echinocandins [see Warnings and Precautions (5.2)]
- •
- Patients with known or suspected Hereditary Fructose Intolerance (HFI) [see Warnings and Precautions (5.4)]
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Hepatic Adverse Reactions
Laboratory abnormalities in liver tests have been seen in healthy volunteers and pediatric patients treated with ERAXIS. In some patients with serious underlying medical conditions who were receiving multiple concomitant medications along with ERAXIS, clinically significant hepatic abnormalities have occurred. Isolated cases of significant hepatic dysfunction, hepatitis, or hepatic failure have been reported in patients; a causal relationship to ERAXIS has not been established [see Adverse Reactions (6.1) and Nonclinical Toxicology (13.2)]. Patients who develop abnormal liver tests during ERAXIS therapy should be monitored for evidence of worsening hepatic tests and evaluated for risk/benefit of continuing ERAXIS therapy.
5.2 Anaphylactic and Hypersensitivity Reactions
Anaphylactic reactions, including shock were reported with the use of ERAXIS. If these reactions occur, ERAXIS should be discontinued and appropriate treatment administered [see Adverse Reactions (6)].
Infusion-related adverse reactions, possibly histamine-mediated, have been reported with ERAXIS, including rash, urticaria, flushing, pruritus, bronchospasm, dyspnea, and hypotension [see Adverse Reactions (6)]. To reduce occurrence of these reactions, do not exceed a rate of ERAXIS infusion of 1.1 mg/minute [see Dosage and Administration (2.4)].
5.3 Risk of Neonatal Toxicity Associated with Polysorbates
ERAXIS contains polysorbate 80, an inactive ingredient. Thrombocytopenia, renal dysfunction, hepatomegaly, cholestasis, ascites, hypotension, and metabolic acidosis have been reported in low-birth weight infants receiving high doses of polysorbate. Polysorbate toxicity has not been reported with ERAXIS. ERAXIS is not approved in pediatric patients younger than 1 month of age [see Indications and Usage (1.1, 1.3), Use in Specific Populations (8.4)].
5.4 Risk in Patients with Hereditary Fructose Intolerance (HFI)
ERAXIS contains fructose, an inactive ingredient, and may precipitate a metabolic crisis that may include, but is not limited to life-threatening hypoglycemia, hypophosphatemia, lactic acidosis, and hepatic failure in patients with HFI. Obtain careful history of HFI symptoms (nausea, vomiting, abdominal pain) with fructose/sucrose exposure prior to ERAXIS administration because a diagnosis of HFI may not yet be established in pediatric patients [see Contraindications (4) and Use in Specific Populations (8.4)].
Adverse Reactions
6 ADVERSE REACTIONS
The following most serious adverse reactions are described elsewhere in other labeling sections:
- •
- Hepatic Adverse Reactions [see Warnings and Precautions (5.1)]
- •
- Anaphylactic and Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials Experience in Adults
The safety of ERAXIS for Injection was assessed in 929 individuals, including 257 healthy subjects and 672 patients in clinical trials of candidemia, other forms of Candida infections, and esophageal candidiasis. A total of 633 patients received ERAXIS at daily doses of either 50 mg or 100 mg. A total of 481 patients received ERAXIS for ≥14 days.
Candidemia/other Candida Infections
Three trials (one comparative vs. fluconazole, two non-comparative) assessed the efficacy and safety of ERAXIS (100 mg) in patients with candidemia and other Candida infections.
The data described below reflect exposure to ERAXIS and fluconazole in 127 and 118 patients, respectively, with candidemia and other forms of invasive candidiasis, in the randomized, comparative trial of the efficacy and safety of ERAXIS to that of fluconazole. In ERAXIS-treated patients, the age range was 16–89 years, the gender distribution was 51% male and 49% female, and the race distribution was 72% White, 18% Black/African American, 9% other races. Patients were randomized to receive once daily IV ERAXIS (200 mg loading dose followed by 100 mg maintenance dose) or IV fluconazole (800 mg loading dose followed by 400 mg maintenance dose). Treatment was administered for at least 14 and not more than 42 days.
The number of patients with adverse reactions leading to discontinuation of study medication was 11.5% in the ERAXIS arm and 21.6% in the fluconazole arm. The most common adverse reactions leading to study drug discontinuation were multi-organ failure (2.3%) and systemic Candida infection (1.5%) in the ERAXIS arm.
Table 2 presents adverse reactions that were reported in ≥5% of subjects receiving ERAXIS or fluconazole therapy in this trial.
| Adverse Reaction | ERAXIS 100 mg N=131 | Fluconazole 400 mg N=125 |
|---|---|---|
| N (%) | N (%) | |
| ||
Subjects with a least one adverse reaction | 130 (99) | 122 (98) |
| ||
Gastrointestinal disorders | 81 (62) | 72 (58) |
Nausea | 32 (24) | 15 (12) |
Diarrhea | 24 (18) | 23 (18) |
Vomiting | 23 (18) | 12 (10) |
Constipation | 11 (8) | 14 (11) |
Abdominal pain | 8 (6) | 16 (13) |
General disorders and administration site conditions | 70 (53) | 76 (61) |
Pyrexia | 23 (18) | 23 (18) |
Edema peripheral | 14 (11) | 16 (13) |
Chest pain | 7 (5) | 6 (5) |
Respiratory, thoracic, and mediastinal disorders | 67 (51) | 55 (44) |
Dyspnea | 15 (12) | 4 (3) |
Pleural effusion | 13 (10) | 11 (9) |
Cough | 9 (7) | 7 (6) |
Respiratory distress | 8 (6) | 2 (2) |
Investigations | 66 (50) | 46 (37) |
Blood alkaline phosphatase increased | 15 (12) | 14 (11) |
White blood cell increased | 11 (8) | 3 (2) |
Hepatic enzyme increased | 7 (5) | 14 (11) |
Blood creatinine increased | 7 (5) | 1 (1) |
Metabolism and nutrition disorders | 61 (47) | 61 (49) |
Hypokalemia | 33 (25) | 24 (19) |
Hypomagnesemia | 15 (12) | 14 (11) |
Hypoglycemia | 9 (7) | 10 (8) |
Hyperkalemia | 8 (6) | 14 (11) |
Hyperglycemia | 8 (6) | 8 (6) |
Dehydration | 8 (6) | 2 (2) |
Vascular disorders | 50 (38) | 41 (33) |
Hypotension | 19 (15) | 18 (14) |
Hypertension | 15 (12) | 5 (4) |
Deep vein thrombosis | 13 (10) | 9 (7) |
Psychiatric disorders | 48 (37) | 45 (36) |
Insomnia | 20 (15) | 12 (10) |
Confusional state | 10 (8) | 10 (8) |
Depression | 8 (6) | 5 (4) |
Blood and lymphatic system disorders | 34 (26) | 36 (29) |
Anemia | 12 (9) | 20 (16) |
Thrombocythemia | 8 (6) | 1 (1) |
Leukocytosis | 7 (5) | 6 (5) |
Skin and subcutaneous tissue disorders | 30 (23) | 32 (26) |
Decubitus ulcer | 7 (5) | 10 (8) |
Nervous system disorders | 27 (21) | 31 (25) |
Headache | 11 (8) | 10 (8) |
Musculoskeletal and connective tissue disorders | 27 (21) | 25 (20) |
Back pain | 7 (5) | 13 (10) |
Esophageal Candidiasis
The data described below reflect exposure to ERAXIS and fluconazole in 300 and 301 patients, respectively, with esophageal candidiasis in a randomized trial comparing the efficacy and safety of ERAXIS to that of oral fluconazole. In ERAXIS-treated patients, the age range was 18–68 years, the gender distribution was 42% male and 58% female and the race distribution was 15% White, 49% Black/African American, 15% Asian, 0.3 % Hispanic, 21% other races. Patients were randomized to receive IV ERAXIS (100 mg on day 1, followed by 50 mg per day) or oral fluconazole (200 mg on day 1, followed by 100 mg per day) for 7 days beyond resolution of symptoms (range, 14–21 days).
Twenty-eight (9%) patients in the ERAXIS arm and 36 (12%) patients in the fluconazole arm had adverse reactions related to study medication. The most common adverse reactions leading to study discontinuation were maculopapular rash (1 patient) for the ERAXIS arm. The most common adverse reactions leading to discontinuation were rash (1 patient) and increased AST (1 patient) for the fluconazole arm.
Table 3 presents adverse reactions that were reported in ≥5% of subjects receiving ERAXIS therapy.
| Adverse Reaction | ERAXIS 50 mg N=300 | Fluconazole 100 mg N=301 |
|---|---|---|
| N (%) | N (%) | |
| ||
Subjects with a least one adverse reaction | 239 (80) | 227 (75) |
| ||
Infections and infestations | 115 (38) | 99 (33) |
Oral candidiasis | 15 (5) | 10 (3) |
Gastrointestinal disorders | 106 (35) | 113 (38) |
Diarrhea | 27 (9) | 26 (9) |
Vomiting | 27 (7) | 30 (10) |
Nausea | 20 (7) | 23 (8) |
Dyspepsia | 20 (7) | 21 (7) |
Blood and lymphatic system disorders | 55 (18) | 50 (17) |
Anemia | 25 (8) | 22 (7) |
Metabolism and nutrition disorders | 50 (17) | 46 (15) |
Hypokalemia | 14 (5) | 17 (6) |
General disorders and administration site condition | 49 (16) | 54 (18) |
Pyrexia | 27 (9) | 28 (9) |
Nervous system disorders | 39 (13) | 36 (12) |
Headache | 25 (8) | 20 (7) |
Less Common Adverse Reactions in Adult Patients with Candidemia/other Candida Infections and Esophageal Candidiasis
The following selected adverse reactions occurred in <2% of patients:
Blood and Lymphatic: coagulopathy, thrombocytopenia
Cardiac: atrial fibrillation, bundle branch block (right), sinus arrhythmia, ventricular extrasystoles
Eye: eye pain, vision blurred, visual disturbance
General and Administration Site: infusion related reaction, peripheral edema, rigors
Hepatobiliary: abnormal liver function tests, cholestasis, hepatic necrosis
Infections: clostridial infection
Investigations: amylase increased, bilirubin increased, CPK increased, electrocardiogram QT prolonged, gamma-glutamyl transferase increased, lipase increased, potassium decreased, prothrombin time prolonged, urea increased
Nervous System: convulsion, dizziness
Respiratory, Thoracic and Mediastinal: cough
Skin and Subcutaneous Tissue: angioneurotic edema, erythema, pruritus, sweating increased, urticaria
Vascular: flushing, hot flushes, thrombophlebitis superficial
Clinical Trials Experience in Pediatric Patients with Candidemia/Invasive Candidiasis
The safety of ERAXIS was investigated in 68 pediatric patients from 1 month to less than 18 years of age with candidemia/invasive candidiasis in a prospective, open-label, non-comparative pediatric trial [see Clinical Studies (14.1)]. Overall, the safety profile of ERAXIS in the pediatric patients 1 month and older was similar to that of adults.
Most Common Adverse Reactions
The most common adverse reactions occurring in ≥5% of pediatric patients receiving ERAXIS therapy in the clinical trial are displayed by body system in Table 4.
| Adverse Reaction | 1 month to <2 years N=19 | 2 to <5 years N=19 | 5 to <18 years N=30 | Overall N=68 |
|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | |
| ||||
Subjects with a least one adverse reaction | 17 (90) | 14 (74) | 24 (80) | 55 (81) |
| ||||
Blood and lymphatic system disorders | 9 (47) | 3 (16) | 4 (13) | 16 (24) |
Anemia | 5 (26) | 2 (11) | 0 | 7 (10) |
Thrombocytopenia | 2 (11) | 1 (5) | 1 (3) | 4 (6) |
Gastrointestinal disorders | 8 (42) | 8 (42) | 12 (40) | 28 (41) |
Diarrhea | 4 (21) | 2 (11) | 5 (17) | 11 (16) |
Vomiting | 4 (21) | 5 (26) | 2 (7) | 11 (16) |
Abdominal pain‡ | 0 | 4 (21) | 2 (7) | 6 (9) |
General disorders and administration site condition | 5 (26) | 6 (32) | 9 (30) | 20 (29) |
Pyrexia | 4 (21) | 3 (16) | 5 (17) | 12 (18) |
Laboratory Investigations | 4 (21) | 4 (21) | 8 (27) | 16 (24) |
Alanine aminotransferase increased | 2 (11) | 2 (11) | 2 (7) | 6 (9) |
Aspartate aminotransferase increased | 2 (11) | 1 (5) | 1 (3) | 4 (6) |
Metabolism and nutrition disorders | 3 (16) | 4 (21) | 6 (20) | 13 (19) |
Hypoglycemia | 1 (5) | 2 (11) | 1 (3) | 4 (6) |
Respiratory, thoracic and mediastinal disorders | 5 (26) | 5 (26) | 5 (17) | 15 (22) |
Epistaxis | 1 (5) | 2 (11) | 3 (10) | 6 (9) |
Skin and subcutaneous tissue disorders | 6 (32) | 5 (26) | 5 (17) | 16 (24) |
Rash§ | 3 (16) | 1 (5) | 2 (7) | 6 (9) |
Other adverse reactions were reported in 2 or more pediatric patients and in less than 5% of the 68 pediatric patients treated with ERAXIS in the clinical trial:
Blood and Lymphatic System Disorders: pancytopenia, thrombocytosis, febrile neutropenia, leukopenia, neutropenia
Eye Disorders: Periorbital edema
Gastrointestinal Disorders: gastroesophageal reflux disease, abdominal distension, nausea, stomatitis, dry mouth
General Disorders and Administrative Site Conditions: chest pain, edema peripheral
Infections and Infestations: bacteremia, device related infection, gastroenteritis, lower respiratory tract infection, upper respiratory tract infection, urinary tract infection
Laboratory Investigations: gamma-glutamyltransferase increased, transaminases increased
Metabolism and Nutrition Disorders: hypocalcemia, hypokalemia, hyponatremia, hypoproteinemia
Musculoskeletal and Connective Tissue Disorders: back pain, pain in extremity
Nervous System Disorders: headache, tremor, seizure
Psychiatric Disorders: agitation
Respiratory, Thoracic and Mediastinal Disorders: hemoptysis
Vascular Disorders: hypotension
6.2 Post-marketing Experience
The following adverse reactions have been identified during post approval use of anidulafungin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune: Anaphylactic shock, anaphylactic reaction, bronchospasm [see Warnings and Precautions (5.2)].
Drug Interactions
7 DRUG INTERACTIONS
7.1 Cyclosporine
Administration of multiple doses of anidulafungin and cyclosporine to healthy subjects resulted in no significant alteration in the steady state pharmacokinetics of either drug. No dosage adjustment of cyclosporine or anidulafungin is needed when the two drugs are co-administered [see Clinical Pharmacology (12.3)].
7.2 Voriconazole
Administration of multiple doses of anidulafungin and voriconazole to healthy subjects resulted in no significant alteration in the steady state pharmacokinetics of either drug. No dosage adjustment of voriconazole or anidulafungin is needed when the two drugs are co-administered [see Clinical Pharmacology (12.3)].
7.3 Tacrolimus
Administration of multiple doses of anidulafungin and a single-dose of tacrolimus to healthy subjects resulted in no significant alteration in the steady state pharmacokinetics of either drug. No dosage adjustment of tacrolimus or anidulafungin is needed when the two drugs are co-administered [see Clinical Pharmacology (12.3)].
7.4 Rifampin
Administration of multiple doses of anidulafungin and rifampin to patients resulted in no significant alteration in the steady state pharmacokinetics of anidulafungin. No dosage adjustment of anidulafungin is needed when it is co-administered with rifampin [see Clinical Pharmacology (12.3)].
7.5 Amphotericin B Liposome for Injection
Administration of multiple doses of anidulafungin and liposomal amphotericin B to patients resulted in no significant alteration in the steady state pharmacokinetics of anidulafungin. No dosage adjustment of anidulafungin is needed when it is co-administered with liposomal amphotericin B [see Clinical Pharmacology (12.3)].
Use in Specific Populations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies, ERAXIS can cause fetal harm when administered to a pregnant woman. There are no available human data on the use of ERAXIS in pregnant women to inform a drug-associated risk of adverse developmental outcomes. In animal reproduction studies fetal toxicity was observed in the presence of maternal toxicity when anidulafungin was administered to pregnant rabbits during organogenesis at 4 times the proposed therapeutic maintenance dose of 100 mg/day on the basis of relative body surface area (see Data). Inform pregnant woman of the risk to the fetus.
The estimated background risk of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20% respectively.
Data
Animal Data
In a combined fertility and embryo-fetal development study in rats dosed with anidulafungin for 4 weeks prior to cohabitation and through cohabitation for males or for 2 weeks prior to cohabitation and continuing through gestation day 19 for females, there was no maternal or embryo-fetal toxicity at intravenous doses up to 20 mg/kg/day (equivalent to 2 times the proposed therapeutic maintenance dose of 100 mg/day on the basis of relative body surface area).
In a rabbit embryo-fetal development study, intravenous administration of anidulafungin (0, 5, 10, and 20 mg/kg/day) from gestation day 7 through 19, resulted in reduced fetal weights and incomplete ossification in the presence of maternal toxicity (decreased body weight gain) at 20 mg/kg/day (equivalent to 4 times the proposed therapeutic maintenance dose of 100 mg/day on the basis of relative body surface area).
In a pre- and postnatal development study, pregnant rats were intravenously administered anidulafungin at doses of 2, 6, or 20 mg/kg/day from gestation day 7 through lactation day 20. Maternal toxicity was observed at ≥6 mg/kg/day (clinical signs at ≥6 mg/kg/day and reduced body weight gain and food consumption during gestation at 20 mg/kg/day group). There were no effects on the viability or growth and development of the offspring. In this study, anidulafungin was detected in fetal plasma, indicating that it crossed the placental barrier.
8.2 Lactation
Risk Summary
There are no data on the presence of anidulafungin in human milk, the effects on the breastfed infant or the effects on milk production. When a drug is present in animal milk, it is likely that the drug will be present in human milk. Anidulafungin was found in the milk of lactating rats (see Data).
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ERAXIS and any potential adverse effects on the breastfed child from ERAXIS or from the underlying maternal condition.
Data
Animal Data
Pregnant rats were intravenously administered anidulafungin at doses of 2, 6, or 20 mg/kg/day from gestation day 7 through lactation day 20. Milk samples were collected from 5 rats per group on lactation day 14 at approximately 1 hours post dose. Approximately dose-proportional anidulafungin concentrations were found in the milk of lactating rats.
8.4 Pediatric Use
The safety and effectiveness of ERAXIS for the treatment of candidemia and the following Candida infections: intra-abdominal abscess and peritonitis, have been established in pediatric patients 1 month of age and older. Use of ERAXIS for this indication in this age group is supported by evidence from adequate and well-controlled studies in adults with additional pharmacokinetic, safety data in pediatric patients 1 month of age and older [see Indications and Usage (1), Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.1)].
The safety and effectiveness of ERAXIS in patients younger than 1 month of age with candidemia/invasive candidiasis has not been established.
Candidemia/invasive candidiasis in pediatric patients younger than 1 month of age has a higher rate of central nervous system (CNS) and multi-organ dissemination than in older patients. In addition, in patients younger than 1 month of age ERAXIS carries a potential risk of life-threatening toxicity associated with high doses of polysorbate 80, an inactive ingredient in ERAXIS [see Warnings and Precautions (5.3)].
The safety and effectiveness of ERAXIS in pediatric patients with esophageal candidiasis has not been established.
ERAXIS is contraindicated in adult and pediatric patients with HFI. Because a diagnosis of HFI may not yet be established in pediatric patients, obtain a careful history of HFI symptoms with fructose/sucrose exposure prior to administration of ERAXIS [see Warnings and Precautions (5.4)].
8.5 Geriatric Use
Of the total number of subjects (N = 197) in the pivotal clinical studies of anidulafungin, 35% were 65 years and over, while 18% were 75 years and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Dosage adjustments are not required for geriatric patients [see Clinical Pharmacology (12.3)].
8.6 Hepatic Insufficiency
No dosing adjustments are required for patients with any degree of hepatic insufficiency. Anidulafungin is not hepatically metabolized. Anidulafungin pharmacokinetics were examined in subjects with Child-Pugh class A, B or C hepatic insufficiency. Anidulafungin concentrations were not increased in subjects with any degree of hepatic insufficiency. Though a slight decrease in AUC was observed in patients with Child-Pugh C hepatic insufficiency, it was within the range of population estimates noted for healthy subjects [see Clinical Pharmacology (12.3)].
8.7 Renal Insufficiency
Dosage adjustments are not required for patients with any degree of renal insufficiency including those on hemodialysis. Anidulafungin has negligible (<1%) renal clearance. In a clinical study of subjects with mild, moderate, severe or end stage (dialysis-dependent) renal insufficiency, anidulafungin pharmacokinetics were similar to those observed in subjects with normal renal function. Anidulafungin is not dialyzable and may be administered without regard to the timing of hemodialysis [see Clinical Pharmacology (12.3)].
Overdosage
10 OVERDOSAGE
During clinical trials a single 400 mg dose of ERAXIS was inadvertently administered as a loading dose. No clinical adverse events were reported. In a study of 10 healthy subjects administered a loading dose of 260 mg followed by 130 mg daily; 3 of the 10 subjects experienced transient, asymptomatic transaminase elevations (≤3 × ULN) [see Warnings and Precautions (5.1)].
Anidulafungin is not dialyzable.
The maximum non-lethal dose of anidulafungin in rats was 50 mg/kg, a dose which is equivalent to 10 times the recommended daily dose for esophageal candidiasis (50 mg/day) or equivalent to 5 times the recommended daily dose for candidemia and other Candida infections (100 mg/day), based on relative body surface area comparisons.
Description
11 DESCRIPTION
ERAXIS for Injection is a sterile, lyophilized product for intravenous (IV) infusion that contains anidulafungin. ERAXIS (anidulafungin) is a semi-synthetic lipopeptide synthesized from a fermentation product of Aspergillus nidulans. Anidulafungin is an echinocandin, a class of antifungal drugs that inhibits the synthesis of 1,3-β-D-glucan, an essential component of fungal cell walls.
ERAXIS (anidulafungin) is 1-[(4R,5R)-4,5-dihydroxy-N2-[[4"-(pentyloxy)[1,1':4',1"-terphenyl]-4-yl]carbonyl]-L-ornithine]echinocandin B. Anidulafungin is a white to off-white powder that is practically insoluble in water and slightly soluble in ethanol. In addition to the active ingredient, anidulafungin, ERAXIS for Injection contains the following inactive ingredients:
50 mg/vial – fructose (50 mg), mannitol (250 mg), polysorbate 80 (125 mg), tartaric acid (5.6 mg), and sodium hydroxide and/or hydrochloric acid for pH adjustment.
100 mg/vial – fructose (100 mg), mannitol (500 mg), polysorbate 80 (250 mg), tartaric acid (11.2 mg), and sodium hydroxide and/or hydrochloric acid for pH adjustment.
The empirical formula of anidulafungin is C58H73N7O17 and the formula weight is 1140.3.
The structural formula is:
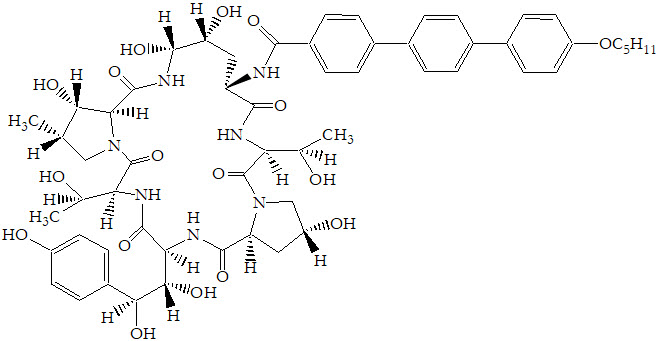
Prior to administration, ERAXIS for Injection requires reconstitution with sterile Water for Injection and subsequent dilution with either 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP (normal saline).
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.3 Pharmacokinetics
General Pharmacokinetic Characteristics
The pharmacokinetics of anidulafungin following intravenous (IV) administration of ERAXIS have been characterized in healthy subjects, special populations and patients. Systemic exposures of anidulafungin are dose-proportional and have low inter-subject variability (coefficient of variation <25%) as shown in Table 5. The steady state was achieved on the first day after a loading dose (twice the daily maintenance dose) and the estimated plasma accumulation factor at steady state is approximately 2.
| Anidulafungin IV Dosing Regimen (LD/MD, mg)* | |||
|---|---|---|---|
| PK Parameter† | 70/35‡,§ (N=6) | 200/100 (N=10) | 260/130§,¶ (N=10) |
| Cmax, ss = the steady state peak concentration | |||
| AUCss = the steady state area under concentration vs. time curve | |||
| CL = clearance | |||
| t1/2 = the terminal elimination half-life | |||
Cmax, ss [mg/L] | 3.55 (13.2) | 8.6 (16.2) | 10.9 (11.7) |
AUCss [mg∙h/L] | 42.3 (14.5) | 111.8 (24.9) | 168.9 (10.8) |
CL [L/h] | 0.84 (13.5) | 0.94 (24.0) | 0.78 (11.3) |
t1/2 [h] | 43.2 (17.7) | 52.0 (11.7) | 50.3 (9.7) |
The clearance of anidulafungin is about 1 L/h and anidulafungin has a terminal elimination half-life of 40–50 hours.
Distribution
The pharmacokinetics of anidulafungin following IV administration are characterized by a short distribution half-life (0.5–1 hour) and a volume of distribution of 30–50 L that is similar to total body fluid volume. Anidulafungin is extensively bound (>99%) to human plasma proteins.
Metabolism
Hepatic metabolism of anidulafungin has not been observed. Anidulafungin is not a clinically relevant substrate, inducer, or inhibitor of cytochrome P450 (CYP450) isoenzymes. It is unlikely that anidulafungin will have clinically relevant effects on the metabolism of drugs metabolized by CYP450 isoenzymes.
Anidulafungin undergoes slow chemical degradation at physiologic temperature and pH to a ring-opened peptide that lacks antifungal activity. The in vitro degradation half-life of anidulafungin under physiologic conditions is about 24 hours. In vivo, the ring-opened product is subsequently converted to peptidic degradants and eliminated.
Excretion
In a single-dose clinical study, radiolabeled (14C) anidulafungin was administered to healthy subjects. Approximately 30% of the administered radioactive dose was eliminated in the feces over 9 days, of which less than 10% was intact drug. Less than 1% of the administered radioactive dose was excreted in the urine. Anidulafungin concentrations fell below the lower limits of quantitation 6 days post-dose. Negligible amounts of drug-derived radioactivity were recovered in blood, urine, and feces 8 weeks post-dose.
Specific Populations
Patients with fungal infections
Population pharmacokinetic analyses from four clinical trials including 107 male and 118 female patients with fungal infections showed that the pharmacokinetic parameters of anidulafungin are not affected by age, race, or the presence of concomitant medications which are known metabolic substrates, inhibitors or inducers.
The pharmacokinetics of anidulafungin in patients with fungal infections are similar to those observed in healthy subjects. The pharmacokinetic parameters of anidulafungin estimated using population pharmacokinetic modeling following IV administration of a maintenance dose of 50 mg/day or 100 mg/day (following a loading dose) of ERAXIS are presented in Table 6.
| PK Parameter* | ERAXIS IV Dosing Regimen (LD/MD, mg)† | |
|---|---|---|
| 100/50 | 200/100 | |
| ||
Cmax, ss [mg/L] | 4.2 (22.4) | 7.2 (23.3) |
Cmin, ss [mg/L] | 1.6 (42.1) | 3.3 (41.8) |
AUCss [mg∙h/L] | 55.2 (32.5) | 110.3 (32.5) |
CL [L/h] | 1.0 (33.5) | |
t1/2, β [h] ‡ | 26.5 (28.5) | |
Gender
Dosage adjustments are not required based on gender. Plasma concentrations of anidulafungin in healthy men and women were similar. In multiple-dose patient studies, drug clearance was slightly faster (approximately 22%) in men.
Geriatric
Dosage adjustments are not required for geriatric patients. The population pharmacokinetic analysis showed that median clearance differed slightly between the elderly group (patients ≥65, median CL=1.07 L/h) and the non-elderly group (patients <65, median CL=1.22 L/h) and the range of clearance was similar.
Race
Dosage adjustments are not required based on race. Anidulafungin pharmacokinetics were similar among Whites, Blacks, Asians, and Hispanics.
HIV Status
Dosage adjustments are not required based on HIV status, irrespective of concomitant anti-retroviral therapy.
Hepatic Insufficiency
Anidulafungin is not hepatically metabolized. Anidulafungin pharmacokinetics were examined in subjects with Child-Pugh class A, B or C hepatic insufficiency. Anidulafungin concentrations were not increased in subjects with any degree of hepatic insufficiency. Though a slight decrease in AUC was observed in patients with Child-Pugh C hepatic insufficiency, it was within the range of population estimates noted for healthy subjects [see Use in Specific Populations (8.6)].
Renal Insufficiency
Anidulafungin has negligible renal clearance. In a clinical study of subjects with mild, moderate, severe or end stage (dialysis-dependent) renal insufficiency, anidulafungin pharmacokinetics were similar to those observed in subjects with normal renal function. Anidulafungin is not dialyzable and may be administered without regard to the timing of hemodialysis [see Use in Specific Populations (8.7)].
Pediatric
The pharmacokinetics of anidulafungin after daily doses were investigated in immunocompromised pediatric (2 through 11 years) and adolescent (12 through 17 years) patients with neutropenia. The steady state was achieved on the first day after administration of the loading dose (twice the maintenance dose), and the Cmax and AUCss increased in a dose-proportional manner. Concentrations and exposures following administration of maintenance doses of 0.75 and 1.5 mg/kg/day in this population were similar to those observed in adults following maintenance doses of 50 and 100 mg/day, respectively (as shown in Table 7).
PK Parameter* | ERAXIS IV Dosing Regimen (LD/MD, mg/kg)† | |||
|---|---|---|---|---|
| 1.5/0.75 | 3.0/1.5 | |||
| Age Group | 2–11 years (N = 6) | 12–17 years (N = 6) | 2–11 years (N = 6) | 12–17 years (N = 6) |
Cmax, ss [mg/L] | 3.32 (50.0) | 4.35 (22.5) | 7.57 (34.2) | 6.88 (24.3) |
AUCss [mg∙h/L] | 41.1 (38.4) | 56.2 (27.8) | 96.1 (39.5) | 102.9 (28.2) |
The pharmacokinetics of anidulafungin were also investigated in 66 pediatric patients (1 month to <18 years) with candidemia/invasive candidiasis (ICC) in a prospective, open-label, non-comparative pediatric study following administration of ERAXIS of 3 mg/kg loading dose on Day 1 and followed by 1.5 mg/kg once daily maintenance dose [see Clinical Studies (14.1)]. Based on population pharmacokinetic analysis of combined data from adult and pediatric patients with ICC, the mean steady state exposure parameters (AUC0–24,,ss Cmin,,ss and Cmax,ss) across age groups in the overall pediatric patients (Table 8) were comparable to those in adults receiving 200 mg loading dose and 100 mg once daily maintenance dose.
| PK Parameter | ERAXIS IV Dosing Regimen (3 mg/kg LD/1.5 mg/kg MD)* | ||
|---|---|---|---|
| Age Group | 1 month to <2 years (N = 17) | 2 to < 5 years (N = 19) | 5 to <18 years (N = 30) |
| |||
Cmax, ss [mg/L] | 6.7 (28.3) | 7.1 (39.4) | 6.5 (29.2) |
Cmin, ss [mg/L] | 2.0 (29) | 2.5 (44) | 2.5 (38.4) |
AUCss [mg∙h/L] | 69.9 (25.3) | 82.8 (38.5) | 86.8 (35.8) |
Drug Interactions
In vitro studies showed that anidulafungin is not metabolized by human cytochrome P450 or by isolated human hepatocytes and does not significantly inhibit the activities of human CYP isoforms (1A2, 2B6, 2C8, 2C9, 2C19, 2D6 and 3A) in clinically relevant concentrations. No clinically relevant drug-drug interactions were observed with drugs likely to be co-administered with anidulafungin.
Cyclosporine (CYP3A4 substrate)
In a study in which 12 healthy adult subjects received 100 mg/day maintenance dose of anidulafungin following a 200 mg loading dose (on Days 1 to 8) and in combination with 1.25 mg/kg oral cyclosporine twice daily (on Days 5 to 8), the steady state Cmax of anidulafungin was not significantly altered by cyclosporine; the steady state AUC of anidulafungin was increased by 22%. A separate in vitro study showed that anidulafungin has no effect on the metabolism of cyclosporine [see Drug Interactions (7.1)].
Voriconazole (CYP2C19, CYP2C9, CYP3A4 inhibitor and substrate)
In a study in which 17 healthy subjects received 100 mg/day maintenance dose of anidulafungin following a 200 mg loading dose, 200 mg twice daily oral voriconazole (following two 400 mg loading doses) and both in combination, the steady state Cmax and AUC of anidulafungin and voriconazole were not significantly altered by co-administration [see Drug Interactions (7.2)].
Tacrolimus (CYP3A4 substrate)
In a study in which 35 healthy subjects received a single oral dose of 5 mg tacrolimus (on Day 1), 100 mg/day maintenance dose of anidulafungin following a 200 mg loading dose (on Days 4 to 12) and both in combination (on Day 13), the steady state Cmax and AUC of anidulafungin and tacrolimus were not significantly altered by co-administration [see Drug Interactions (7.3)].
Rifampin (potent CYP450 inducer)
The pharmacokinetics of anidulafungin were examined in 27 patients that were co-administered anidulafungin and rifampin. The population pharmacokinetic analysis showed that when compared to data from patients that did not receive rifampin, the pharmacokinetics of anidulafungin were not significantly altered by co-administration with rifampin [see Drug Interactions (7.4)].
Amphotericin B liposome for injection
The pharmacokinetics of anidulafungin were examined in 27 patients that were co-administered liposomal amphotericin B. The population pharmacokinetic analysis showed that when compared to data from patients that did not receive amphotericin B, the pharmacokinetics of anidulafungin were not significantly altered by co-administration with amphotericin B [see Drug Interactions (7.5)].
12.4 Microbiology
Mechanism of Action
Anidulafungin is a semi-synthetic echinocandin with antifungal activity. Anidulafungin inhibits glucan synthase, an enzyme present in fungal, but not mammalian cells. This results in inhibition of the formation of 1,3-β-D-glucan, an essential component of the fungal cell wall.
Resistance
Echinocandin resistance is due to point mutations within the genes (FKS1 and FKS2) encoding for subunits in the glucan synthase enzyme complex. There have been reports of Candida isolates with reduced susceptibility to anidulafungin, suggesting a potential for development of drug resistance. The clinical significance of this observation is not fully understood.
Antimicrobial Activity
Anidulafungin has been shown to be active against most isolates of the following microorganisms both in vitro and in clinical infections:
Candida albicans
Candida glabrata
Candida parapsilosis
Candida tropicalis
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following fungi exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for anidulafungin against isolates of the following Candida species. However, the effectiveness of anidulafungin in treating clinical infections due to these fungi has not been established in adequate and well-controlled clinical trials:
Candida guilliermondii
Candida krusei
Nonclinical Toxicology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal carcinogenicity studies of anidulafungin have not been conducted.
Anidulafungin was not genotoxic in the following in vitro studies: bacterial reverse mutation assays, a chromosome aberration assay with Chinese hamster ovary cells, and a forward gene mutation assay with mouse lymphoma cells. Anidulafungin was not genotoxic in mice using the in vivo micronucleus assay.
Anidulafungin produced no adverse effects on fertility in male or female rats at intravenous doses of 20 mg/kg/day (equivalent to 2 times the proposed therapeutic maintenance dose of 100 mg/day on the basis of relative body surface area).
13.2 Animal Toxicology and/or Pharmacology
In 3 month studies, liver toxicity, including single cell hepatocellular necrosis, hepatocellular hypertrophy and increased liver weights were observed in monkeys and rats at doses equivalent to 5–6 times human exposure. For both species, hepatocellular hypertrophy was still noted one month after the end of dosing.
Clinical Studies
14 CLINICAL STUDIES
14.1 Candidemia and Other Candida Infections (Intra-abdominal Abscess and Peritonitis)
Adults Candidemia and Other Candida Infections (Intra-abdominal Abscess and Peritonitis) Trial
The safety and efficacy of ERAXIS were evaluated in a Phase 3, randomized, double-blind study of patients with candidemia and/or other forms of invasive candidiasis. Patients were randomized to receive once daily IV ERAXIS (200 mg loading dose followed by 100 mg maintenance dose) or IV fluconazole (800 mg loading dose followed by 400 mg maintenance dose). Patients were stratified by APACHE II score (≤20 and >20) and the presence or absence of neutropenia. Patients with Candida endocarditis, osteomyelitis or meningitis, or those with infection due to C. krusei, were excluded from the study. Treatment was administered for at least 14 and not more than 42 days. Patients in both study arms were permitted to switch to oral fluconazole after at least 10 days of intravenous therapy, provided that they were able to tolerate oral medication, were afebrile for at least 24 hours, and the last blood cultures were negative for Candida species.
Patients who received at least one dose of study medication and who had a positive culture for Candida species from a normally sterile site before entry into the study (modified intent-to-treat [MITT] population) were included in the analysis of global response at the end of IV therapy. A successful global response required clinical cure or improvement (significant, but incomplete resolution of signs and symptoms of the Candida infection and no additional antifungal treatment), and documented or presumed microbiological eradication. Patients with an indeterminate outcome were analyzed as failures in this population.
Two hundred and fifty-six patients in the intent-to-treat (ITT) population were randomized and received at least one dose of study medication. In ERAXIS-treated patients, the age range was 16–89 years, the gender distribution was 50% male and 50% female, and the race distribution was 71% White, 20% Black/African American, 7% Hispanic, 2% other races. The median duration of IV therapy was 14 and 11 days in the ERAXIS and fluconazole arms, respectively. For those who received oral fluconazole, the median duration of oral therapy was 7 days for the ERAXIS arm and 5 days for the fluconazole arm.
Patient disposition is presented in Table 9.
| ERAXIS | Fluconazole | |
|---|---|---|
| n (%) | n (%) | |
Treated patients | 131 | 125 |
Patients completing study through 6 week follow-up | 94 (72) | 80 (64) |
DISCONTINUATIONS FROM STUDY MEDICATION | ||
Total discontinued from study medication | 34 (26) | 48 (38) |
Discontinued due to adverse events | 12 (9) | 21 (17) |
Discontinued due to lack of efficacy | 11 (8) | 16 (13) |
Two hundred and forty-five patients (127 ERAXIS, 118 fluconazole) met the criteria for inclusion in the MITT population. Of these, 219 patients (116 ERAXIS, 103 fluconazole) had candidemia only. Risk factors for candidemia among patients in both treatment arms in this study were: presence of a central venous catheter (78%), receipt of broad-spectrum antibiotics (69%), recent surgery (42%), recent hyperalimentation (25%), and underlying malignancy (22%). The most frequent species isolated at baseline was C. albicans (62%), followed by C. glabrata (20%), C. parapsilosis (12%) and C. tropicalis (11%). The majority (97%) of patients were non-neutropenic (ANC >500) and 81% had APACHE II scores less than or equal to 20.
Global success rates in patients with candidemia and other Candida infections are summarized in Table 10.
| Time-point | ERAXIS (N=127) n (%) | Fluconazole (N=118) n (%) | Treatment Difference*, % (95% C.I.) |
|---|---|---|---|
End of IV Therapy | 96 (75.6) | 71 (60.2) | 15.4 |
End of All Therapy† | 94 (74.0) | 67 (56.8) | 17.2 |
2 Week Follow-up | 82 (64.6) | 58 (49.2) | 15.4 |
6 Week Follow-up | 71 (55.9) | 52 (44.1) | 11.8 |
Table 11 presents global response by patients with candidemia or multiple sites of Candida infection and mortality data for the MITT population.
| ERAXIS | Fluconazole | Between group difference * (95% CI) | |
|---|---|---|---|
| |||
No. of MITT patients | 127 | 118 | |
Global Success (MITT) At End Of IV Therapy | |||
| |||
Candidemia | 88/116 (75.9%) | 63/103 (61.2%) | 14.7 (2.5, 26.9) |
Neutropenic | 1/2 | 2/4 | - |
Non neutropenic | 87/114 (76.3%) | 61/99 (61.6%) | - |
| |||
Multiple sites | |||
Peritoneal fluid/ intra-abdominal abscess | 4/6 | 5/6 | - |
Blood/ peritoneum (intra-abdominal abscess) | 2/2 | 0/2 | - |
Blood /bile | - | 1/1 | - |
Blood/renal | - | 1/1 | - |
Pancreas | - | 0/3 | - |
Pelvic abscess | - | 1/2 | - |
Pleural fluid | 1/1 | - | - |
Blood/ pleural fluid | 0/1 | - | - |
Blood/left thigh lesion biopsy | 1/1 | - | - |
Total | 8/11 (72.7%) | 8/15 (53.3%) | - |
Mortality | |||
Overall study mortality | 29/127 (22.8 %) | 37/118 (31.4%) | - |
Mortality during study therapy | 10/127 (7.9%) | 17/118 (14.4%) | - |
Mortality attributed to Candida | 2/127 (1.6%) | 5/118 (4.2%) | - |
Pediatric Candidemia/Invasive Candidiasis Trial
A prospective, open-label, non-comparative, multinational trial (National Clinical Trial (NCT) number 00761267) assessed the safety and efficacy of ERAXIS in 68 pediatric patients aged 1 month to <18 years with candidemia/invasive candidiasis. Patients were stratified by age (1 month to <2 years, 2 to <5 years, and 5 to <18 years) and received once daily intravenous ERAXIS (3 mg/kg loading dose on Day 1, and 1.5 mg/kg daily maintenance dose thereafter) followed by an optional switch to oral fluconazole (6–12 mg/kg/day, maximum 800 mg/day) up to Day 49. Patients were followed at 2 and 6 weeks after EOT.
Among 68 ERAXIS-treated patients, 64 had microbiologically confirmed Candida infection and were evaluated for efficacy in the modified intent-to-treat (MITT) population. Overall, 59 patients (92.2%) had Candida isolated from blood only. The most commonly isolated pathogens were Candida albicans (25 [39.1%] patients), followed by Candida parapsilosis (17 [26.6%] patients), and Candida tropicalis (9 [14.1%] patients). A successful global response was defined as having both successful clinical (cure or improvement) and microbiological (eradication or presumed eradication) response. The overall rates of successful global response in the MITT population are presented in Table 12 below.
| Successful Global Response, n (%) | |||||
|---|---|---|---|---|---|
| Timepoint | Global Response | 1 month to <2 years (N = 16) n (n/N, %) | 2 to <5 years (N=18) n (n/N, %) | 5 to <18 years (N=30) n (n/N, %) | Overall (N=64) n (n/N, %) |
| 95% CI = exact 95% confidence interval for binomial proportions using Clopper-Pearson method; EOIVT = end of intravenous treatment; EOT = end of treatment; FU = follow-up; MITT = modified intent-to-treat; N = number of subjects in the population; n = number of subjects with responses. | |||||
EOIVT | Success | 11 (68.8) | 14 (77.8) | 20 (66.7) | 45 (70.3) |
| (41.3, 89.0) | (52.4, 93.6) | (47.2, 82.7) | (57.6, 81.1) | |
EOT | Success | 11 (68.8) | 14 (77.8) | 21 (70.0) | 46 (71.9) |
| (41.3, 89.0) | (52.4, 93.6) | (50.6, 85.3) | (59.2, 82.4) | |
2-week FU | Success | 11 (68.8) | 13 (72.2) | 22 (73.3) | 46 (71.9) |
| (41.3, 89.0) | (46.5, 90.3) | (54.1, 87.7) | (59.2, 82.4) | |
6-week FU | Success | 11 (68.8) | 12 (66.7) | 20 (66.7) | 43 (67.2) |
| (41.3, 89.0) | (41.0, 86.7) | (47.2, 82.7) | (54.3, 78.4) | |
14.2 Esophageal Candidiasis
ERAXIS was evaluated in a double-blind, double-dummy, randomized Phase 3 study. Three hundred patients received ERAXIS (100 mg loading dose IV on Day 1 followed by 50 mg/day IV) and 301 received oral fluconazole (200 mg loading dose on Day 1 followed by 100 mg/day). Treatment duration was 7 days beyond resolution of symptoms for a minimum of 14 and a maximum of 21 days.
Of the 442 patients with culture confirmed esophageal candidiasis, most patients (91%) had C. albicans isolated at the baseline.
Treatment groups were similar in demographic and other baseline characteristics. In ERAXIS-treated patients, the age range was 16–69 years, the gender distribution was 42% male and 58% female, and the race distribution was 15% White, 49% Black/African American, 15% Asian, 0.3 % Hispanic, 21% other races.
In this study, of 280 patients tested, 237 (84.6%) tested HIV positive. In both groups the median time to resolution of symptoms was 5 days and the median duration of therapy was 14 days.
Efficacy was assessed by endoscopic outcome at end of therapy (EOT). Patients were considered clinically evaluable if they received at least 10 days of therapy, had an EOT assessment with a clinical outcome other than 'indeterminate', had an endoscopy at EOT, and did not have any protocol violations prior to the EOT visit that would affect an assessment of efficacy.
An endoscopic success, defined as cure (endoscopic grade of 0 on a 4-point severity scale) or improvement (decrease of one or more grades from baseline), was seen in 225/231 (97.4%) ERAXIS-treated patients and 233/236 (98.7%) fluconazole-treated patients (Table 13). The majority of these patients were endoscopic cures (grade=0). Two weeks after completing therapy, the ERAXIS group had significantly more endoscopically-documented relapses than the fluconazole group, 120/225 (53.3%) vs. 45/233 (19.3%), respectively (Table 13).
| |||||
Endoscopic Response at End of Therapy | |||||
Response | ERAXIS | Fluconazole | Treatment Difference* | 95% CI | |
Endoscopic Success, n (%) | 225 (97.4) | 233 (98.7) | -1.3% | -3.8%, 1.2% | |
Cure | 204 (88.3) | 221 (93.6) | |||
Improvement | 21 (9.1) | 12 (5.1) | |||
Failure, n (%) | 6 (2.6) | 3 (1.3) | |||
| |||||
Endoscopic Relapse Rates at Follow-Up, 2 Weeks Post-Treatment | |||||
ERAXIS | Fluconazole | Treatment Difference* | 95% CI | ||
Endoscopic Relapse, n/N (%) | 120/225 (53.3%) | 45/233 (19.3%) | 34.0% | 25.8%, 42.3% | |
Clinical success (cure or improvement in clinical symptoms including odynophagia/dysphagia and retrosternal pain) occurred in 229/231 (99.1%) of the ERAXIS-treated patients and 235/236 (99.6%) of the fluconazole-treated patients at the end of therapy. For patients with C. albicans, microbiological success occurred in 142/162 (87.7%) of the ERAXIS-treated group and 157/166 (94.6%) of the fluconazole-treated group at the end of therapy. For patients with Candida species other than C. albicans, success occurred in 10/12 (83.3%) of the ERAXIS-treated group and 14/16 (87.5%) of the fluconazole-treated group.
How Supplied/Storage and Handling
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
ERAXIS (anidulafungin) for Injection is supplied in a single-dose vial of sterile, lyophilized, preservative-free, white to off-white powder. ERAXIS (anidulafungin) is available in the following packaging configuration:
Single-Dose Vial of ERAXIS 50 mg | |
NDC 0049-0114-28 | One - 50 mg vial |
Single-Dose Vial of ERAXIS 100 mg | |
NDC 0049-0116-28 | One - 100 mg vial |
16.2 Storage
ERAXIS vials
ERAXIS (unreconstituted) vials should be stored in a refrigerator at 2°C – 8°C (36°F – 46°F). Do not freeze.
Excursions for 96 hours up to 25°C (77°F) are permitted, and the vial can be returned to storage at 2°C – 8°C (36°F – 46°F).
Reconstituted solution
ERAXIS reconstituted solution can be stored at up to 25°C (77°F) for up to 24 hours [see Dosage and Administration (2.3)].
Infusion Solution
ERAXIS infusion solution can be stored at temperatures up to 25°C (77°F) for up to 48 hours. Do not freeze [see Dosage and Administration (2.4)].
Medication Guide
17 PATIENT COUNSELING INFORMATION
Hepatic Adverse Reactions
Inform patients about the risk of developing abnormal liver function tests and/or hepatic dysfunction. Advise the patient that liver function tests may be monitored during treatment.
Anaphylactic and Hypersensitivity Reactions
Inform the patient that anaphylactic reactions, including shock were reported with ERAXIS. Inform the patient if these reactions occur, ERAXIS may be discontinued and appropriate treatment administered.
Inform the patient that ERAXIS is also known to cause infusion-related adverse reactions, possibly histamine-mediated. Inform the patient to report symptoms including rash, urticaria, flushing, pruritus, dyspnea, and hypotension to their healthcare provider.
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk of ERAXIS to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy.
Patients with Hereditary Fructose Intolerance (HFI)
Inform patients and caregivers that ERAXIS contains fructose and can be life-threatening when administered to patients with hereditary fructose intolerance (HFI) [see Warnings and Precautions (5.4)]. Inquire for symptoms of fructose and/or sucrose intolerance.
Other
This product's prescribing information may have been updated. For current full prescribing information, please visit www.pfizer.com.
For medical information about ERAXIS, please visit www.pfizermedinfo.com or call 1-800-438-1985.

LAB-0336-16.0
17 PATIENT COUNSELING INFORMATION
17 PATIENT COUNSELING INFORMATION
Hepatic Adverse Reactions
Inform patients about the risk of developing abnormal liver function tests and/or hepatic dysfunction. Advise the patient that liver function tests may be monitored during treatment.
Anaphylactic and Hypersensitivity Reactions
Inform the patient that anaphylactic reactions, including shock were reported with ERAXIS. Inform the patient if these reactions occur, ERAXIS may be discontinued and appropriate treatment administered.
Inform the patient that ERAXIS is also known to cause infusion-related adverse reactions, possibly histamine-mediated. Inform the patient to report symptoms including rash, urticaria, flushing, pruritus, dyspnea, and hypotension to their healthcare provider.
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk of ERAXIS to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy.
Patients with Hereditary Fructose Intolerance (HFI)
Inform patients and caregivers that ERAXIS contains fructose and can be life-threatening when administered to patients with hereditary fructose intolerance (HFI) [see Warnings and Precautions (5.4)]. Inquire for symptoms of fructose and/or sucrose intolerance.
Full Patient Information
Full Patient Information
17 PATIENT COUNSELING INFORMATION
Hepatic Adverse Reactions
Inform patients about the risk of developing abnormal liver function tests and/or hepatic dysfunction. Advise the patient that liver function tests may be monitored during treatment.
Anaphylactic and Hypersensitivity Reactions
Inform the patient that anaphylactic reactions, including shock were reported with ERAXIS. Inform the patient if these reactions occur, ERAXIS may be discontinued and appropriate treatment administered.
Inform the patient that ERAXIS is also known to cause infusion-related adverse reactions, possibly histamine-mediated. Inform the patient to report symptoms including rash, urticaria, flushing, pruritus, dyspnea, and hypotension to their healthcare provider.
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk of ERAXIS to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy.
Patients with Hereditary Fructose Intolerance (HFI)
Inform patients and caregivers that ERAXIS contains fructose and can be life-threatening when administered to patients with hereditary fructose intolerance (HFI) [see Warnings and Precautions (5.4)]. Inquire for symptoms of fructose and/or sucrose intolerance.
Resources
Didn’t find what you were looking for?
Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5Pm ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this product, click the link below to submit your information: Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer product, please call Pfizer Medical Information at (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800)-332-1088.