CYKLOKAPRON®
(tranexamic acid)
Find CYKLOKAPRON® medical information:
Find CYKLOKAPRON® medical information:
CYKLOKAPRON® Quick Finder
Highlights
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use CYKLOKAPRON safely and effectively. See full prescribing information for CYKLOKAPRON. CYKLOKAPRON (tranexamic acid) injection, for intravenous use Initial U.S. Approval: 1986 INDICATIONS AND USAGECYKLOKAPRON is an antifibrinolytic indicated in patients with hemophilia for short-term use (2 to 8 days) to reduce or prevent hemorrhage and reduce the need for replacement therapy during and following tooth extraction. (1) DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHSCONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions are nausea, vomiting, diarrhea, allergic dermatitis, giddiness, hypotension, and thromboembolic events. (6) To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 1/2024 |
Indications and Usage
Dosage and Administration
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dose of CYKLOKAPRON is 10 mg/kg actual body weight intravenously administered as a single dose, immediately before tooth extractions Infuse no more than 1 mL/minute to avoid hypotension [see Warnings and Precautions (5.1)]. Following tooth extraction, CYKLOKAPRON may be administered for 2 to 8 days at a dose of 10 mg/kg actual body weight 3 to 4 times daily, intravenously.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
For intravenous infusion, CYKLOKAPRON Injection may be mixed with most solutions for infusion such as electrolyte solutions, carbohydrate solutions, amino acid solutions, and Dextran solutions. Heparin may be added to CYKLOKAPRON Injection. CYKLOKAPRON Injection should NOT be mixed with blood. The drug is a synthetic amino acid and should NOT be mixed with solutions containing penicillin.
Discard any unused portion.
The diluted mixture may be stored for up to 4 hours at room temperature prior to patient administration.
2.2 Recommended Dosage for Patients With Varying Degrees of Renal Impairment*
For patients with moderate to severe impaired renal function, the following dosages are recommended:
| Serum Creatinine (mg/dL) | CYKLOKAPRON Intravenous Dosage |
|---|---|
| * Dose reduction is recommended for all doses, both before and after tooth extraction. | |
1.36 to 2.83 | 10 mg/kg twice daily |
2.83 to 5.66 (250 to 500 micromol/L) | 10 mg/kg daily |
>5.66 (>500 micromol/L) | 10 mg/kg every 48 hours |
Dosage Forms and Strengths
Contraindications
4 CONTRAINDICATIONS
CYKLOKAPRON Injection is contraindicated:
- •
- In patients with subarachnoid hemorrhage. Anecdotal experience indicates that cerebral edema and cerebral infarction may be caused by CYKLOKAPRON in such patients.
- •
- In patients with active intravascular clotting [see Warnings and Precautions (5.1)].
- •
- In patients with hypersensitivity to tranexamic acid or any of the ingredients [see Warnings and Precautions (5.4)].
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Thromboembolic Risk
CYKLOKAPRON is contraindicated in patients with active intravascular clotting.
Tranexamic acid is an antifibrinolytic and may increase the risk of thromboembolic events. Venous and arterial thrombosis or thromboembolism has been reported in patients treated with CYKLOKAPRON. Avoid concomitant use of CYKLOKAPRON and medical products that are pro-thrombotic, as the risk of thrombosis may be increased. These medications include but are not limited to, Factor IX Complex concentrates, Anti-inhibitor Coagulant concentrates, and hormonal contraceptives [see Drug Interactions (7.1), Use in Specific Populations (8.3)].
5.2 Risk of Medication Errors Due to Incorrect Route of Administration
CYKLOKAPRON is for intravenous use only. Serious adverse reactions including seizures and cardiac arrythmias have occurred when CYKLOKAPRON was inadvertently administered intrathecally instead of intravenously.
Confirm the correct route of administration for CYKLOKAPRON and avoid confusion with other injectable solutions that might be administered at the same time as CYKLOKAPRON. Syringes containing CYKLOKAPRON should be clearly labeled with the intravenous route of administration.
5.3 Seizures
CYKLOKAPRON may cause seizures, including focal and generalized seizures. The most common setting for tranexamic acid-induced seizures has been during cardiovascular surgery (a setting in which CYKLOKAPRON is not FDA-approved and which uses doses of up to 10-fold higher than the recommended human dose and in patients inadvertently given tranexamic acid into the neuraxial system). CYKLOKAPRON is not approved and not recommended for neuraxial administration. Consider dose reduction during surgery and dose adjustments for patients with clinical conditions such as renal dysfunction. Closely monitor the patient during surgery. Consider electroencephalogram (EEG) monitoring for patients with history of seizures or who experience myoclonic movements, twitching, or show evidence of focal seizures. Discontinue CYKLOKAPRON if seizures occur.
5.4 Hypersensitivity Reactions
Cases of hypersensitivity reactions, including anaphylactic reactions, have occurred with use of intravenous tranexamic acid. Discontinue treatment with CYKLOKAPRON if serious reaction occurs, provide appropriate medical management, and do not restart treatment. CYKLOKAPRON is contraindicated in patients with a history of hypersensitivity to tranexamic acid.
5.5 Visual Disturbances
Although not seen in humans, focal areas of retinal degeneration have been observed in cats and dogs following oral or intravenous tranexamic acid at doses between 250 to 1600 mg/kg/day (1.6 to 22 times the recommended usual human dose based on body surface area) from 6 days to 1 year. No retinal changes have been observed in eye examinations of patients treated with tranexamic acid for up to 8 years. Patients expected to be treated for greater than 3 months may consider ophthalmic monitoring including visual acuity and optical coherence tomography at regular intervals.
Discontinue CYKLOKAPRON if changes in ophthalmological examination occurs.
Adverse Reactions
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- •
- Thromboembolic Risk [see Warnings and Precautions (5.1)]
- •
- Seizures [see Warnings and Precautions (5.3)]
- •
- Hypersensitivity Reactions [see Warnings and Precautions (5.4)]
- •
- Visual Disturbances [see Warnings and Precautions (5.5)]
- •
- Dizziness [see Warnings and Precautions (5.6)]
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of tranexamic acid. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal disturbances (nausea, vomiting, diarrhea) may occur and may resolve with dose-reduction. Allergic dermatitis and giddiness have been reported. Hypotension has been reported when intravenous injection is too rapid.
Thromboembolic events (e.g., deep vein thrombosis, pulmonary embolism, cerebral thrombosis, acute renal cortical necrosis, and central retinal artery, vein obstruction and cases associated with concomitant use of combination hormonal contraceptives) have been rarely reported in patients receiving tranexamic acid for indications other than hemorrhage prevention in patients with hemophilia. Convulsion, cromatopsia, and visual impairment have also been reported.
Anaphylaxis or anaphylactoid reactions have been reported that are suggestive of a causal relationship.
Drug Interactions
7 DRUG INTERACTIONS
7.1 Prothrombotic Medical Products
Avoid concomitant use of CYKLOKAPRON with medical products that are prothrombotic because concomitant use can further increase the risk of thromboembolic adverse reactions associated with tranexamic acid [see Warnings and Precautions (5.1), Use in Specific Populations (8.3)].
Use in Specific Populations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from published studies, case series and case reports with tranexamic acid use in pregnant women in the second and third trimester and at the time of delivery have not clarified whether there is a drug-associated risk of miscarriage or adverse maternal or fetal outcomes. There are 2 (0.02%) infant cases with structural abnormalities that resulted in death when tranexamic acid was used during conception or the first trimester of pregnancy; however, due to other confounding factors the risk of major birth defects with use of tranexamic acid during pregnancy is not clear. Tranexamic acid is known to pass the placenta and appears in cord blood at concentrations approximately equal to maternal concentration (see Data).
Reproduction studies performed in mice, rats, and rabbits have not revealed any adverse effects on the fetus due to tranexamic acid administered during organogenesis. Doses examined were multiples of up to 3 times (mouse), 6 times (rat), and 3 times (rabbit) the maximum human dose based on body surface area in the mouse, rat, and rabbit, respectively (see Data).
The estimated background risk for major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in the clinically recognized pregnancies is 2–4% and 15–20%, respectively.
It is not known whether tranexamic acid use in pregnant women may cause a drug-associated risk of miscarriage or adverse maternal or fetal outcomes. For decisions regarding the use of CYKLOKAPRON during pregnancy, the potential risk of CYKLOKAPRON administration on the fetus should always be considered along with the mother's clinical need for CYKLOKAPRON; an accurate risk-benefit evaluation should drive the treating physician's decision.
Data
Human Data
Tranexamic acid passes through the placenta. The concentration in cord blood after an intravenous injection of 10 mg/kg to pregnant women is about 30 mg/L, as high as in the maternal blood.
There were 13 clinical studies that described fetal and/or neonatal functional issues such as low Apgar score, neonatal sepsis, cephalohematoma and 9 clinical studies that discussed alterations to growth including low birth weight and preterm birth at 22–36 weeks of gestation in fetuses and infants exposed to tranexamic acid in-utero.
Animal Data
In embryo-fetal development studies, tranexamic acid was administered to pregnant mice from Gestation day (GD) 6 through GD 12 and rats from GD 9 through GD 14 at daily doses of 0.3 or 1.5 g/kg. There was no evidence of adverse developmental outcomes in mice and rats at multiple of 3 and 6 times the maximum recommended human dose based on body surface area in the mouse and rat, respectively.
In rabbits, tranexamic acid was administered intravenously at doses of 50, 100, or 200 mg/kg/day or orally at doses of 100, 200, or 400 mg/kg/day from GD 6 through GD 18. There was no evidence of adverse developmental outcomes at dose multiples of 2 or 3 times, respectively, the maximum recommended human dose based on body surface area. Intravenous doses of 200 mg/kg/day showed slightly retarded weight gain in pregnant rabbits.
8.2 Lactation
Risk Summary
Published literature reports the presence of tranexamic acid in human milk. There are no data on the effects of tranexamic acid on the breastfed child or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CYKLOKAPRON and any potential adverse effects on the breastfed child from CYKLOKAPRON or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Concomitant use of CYKLOKAPRON, which is an antifibrinolytic, with hormonal contraceptives may increase the risk for thromboembolic adverse reactions. Advise patients to use an effective alternative (nonhormonal) contraceptive method [see Warnings and Precautions (5.1), Drug Interactions (7.1)].
8.4 Pediatric Use
There are limited data concerning the use of CYKLOKAPRON in pediatric patients with hemophilia who are undergoing tooth extraction. The limited data suggest that there are no significant pharmacokinetic differences between adults and pediatric patients.
8.5 Geriatric Use
Clinical studies of CYKLOKAPRON did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
8.6 Renal Impairment
Reduce the dosage of CYKLOKAPRON in patients with renal impairment, based on the patient's serum creatinine [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
Overdosage
10 OVERDOSAGE
Cases of overdosage of CYKLOKAPRON have been reported. Based on these reports, symptoms of overdosage may be gastrointestinal, e.g., nausea, vomiting, diarrhea; hypotensive, e.g., orthostatic symptoms; thromboembolic, e.g., arterial, venous, embolic; neurologic, e.g., visual impairment, convulsions, headache, mental status changes; myoclonus; and rash.
Description
11 DESCRIPTION
Tranexamic acid is trans-4-(aminomethyl)cyclohexanecarboxylic acid, an antifibrinolytic agent. Tranexamic acid is a white crystalline powder. The structural formula is
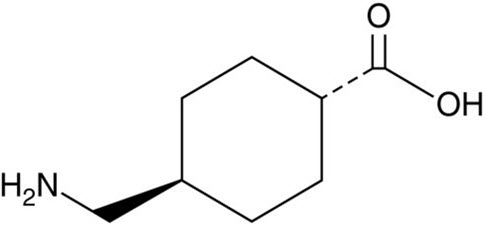
Empirical Formula: C8H15NO2 Molecular Weight: 157.2
Each mL of the sterile solution for intravenous injection contains 100 mg tranexamic acid and Water for Injection to 1 mL. The aqueous solution for injection has a pH of 6.5 to 8.0.
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tranexamic acid is a synthetic lysine amino acid derivative, which diminishes the dissolution of hemostatic fibrin by plasmin. In the presence of tranexamic acid, the lysine receptor binding sites of plasmin for fibrin are occupied, preventing binding to fibrin monomers, thus preserving and stabilizing fibrin's matrix structure.
The antifibrinolytic effects of tranexamic acid are mediated by reversible interactions at multiple binding sites within plasminogen. Native human plasminogen contains 4 to 5 lysine binding sites with low affinity for tranexamic acid (Kd = 750 μmol/L) and 1 with high affinity (Kd = 1.1 μmol/L). The high affinity lysine site of plasminogen is involved in its binding to fibrin. Saturation of the high affinity binding site with tranexamic acid displaces plasminogen from the surface of fibrin. Although plasmin may be formed by conformational changes in plasminogen, binding to and dissolution of the fibrin matrix is inhibited.
12.2 Pharmacodynamics
Tranexamic acid, in concentrations of 1 mg/mL and 10 mg/mL prolongs the thrombin time. An antifibrinolytic concentration of tranexamic acid remains in different tissues for about 17 hours, and in the serum, up to 7 or 8 hours.
Tranexamic acid in concentrations up to 10 mg/mL blood has no influence on the platelet count, the coagulation time or various coagulation factors in whole blood or citrated blood from healthy subjects.
12.3 Pharmacokinetics
Distribution
The initial volume of distribution is about 9 to 12 liters. The plasma protein binding of tranexamic acid is about 3% at therapeutic plasma levels and seems to be fully accounted for by its binding to plasminogen. Tranexamic acid does not bind to serum albumin.
Elimination
After an intravenous dose of 1 g, the plasma concentration time curve shows a triexponential decay with a half-life of about 2 hours for the terminal elimination phase.
Excretion
Urinary excretion is the main route of elimination via glomerular filtration. Overall renal clearance is equal to overall plasma clearance (110 to 116 mL/min), and more than 95% of the dose is excreted in the urine as unchanged drug. Excretion of tranexamic acid is about 90% at 24 hours after intravenous administration of 10 mg/kg body weight.
Specific Populations
Patients with Renal Impairment
The blood levels of tranexamic acid are increased in patients with renal insufficiency. Urinary excretion following a single intravenous injection of tranexamic acid declines as renal function decreases. Following a single 10 mg/kg intravenous injection of tranexamic acid, the 24-hour urinary fractions of tranexamic acid with serum creatinine concentrations 1.4 – 2.8, 2.8 – 5.7, and greater than 5.7 mg/dL were 51, 39, and 19%, respectively. The 24-hour tranexamic acid plasma concentrations for these patients demonstrated a direct relationship to the degree of renal impairment. Therefore, dose adjustment is needed in patients with renal impairment [see Dosage and Administration (2.2), Use in Specific Populations (8.6)].
Nonclinical Toxicology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Tranexamic acid was not carcinogenic in a 2-year study in rats and mice at oral doses up to 3 and 5.3 g/kg/day, which are approximately 12 and 11 times the maximum recommended human dose based on body surface area, respectively.
Tranexamic acid was not genotoxic in the reverse mutation bacterial (Ames) test, and in vitro and in vivo cytogenetic test.
In a fertility and early embryonic development study, tranexamic acid was administered to male rats as 0.3% and 1% of drug in diet (average doses of 222 and 856 mg/kg/day) or to female rats at dose levels of 0.3% and 1.2% of drug in diet. Tranexamic acid had no effect on fertility or reproductive function of male or female rats at dose multiples of 4 or 5 times the maximum recommended human dose based on body surface area, respectively.
13.2 Animal Toxicology and/or Pharmacology
Nonclinical studies have shown a retinal toxicity associated with tranexamic acid. Toxicity is characterized by retinal atrophy commencing with changes to the retinal pigmented epithelium and progressing to retinal detachment in cats. The toxicity appears to be dose related, and changes are partially reversible at lower doses. Effects were observed in dogs at oral doses of 800 mg/kg/day and higher (multiple of 11 times the maximum human dose based on body surface area), and in cats at 250 mg/kg/day for 14 days (multiple of 1.6 times the maximum human dose based on body surface area). Some fully reversible changes in pigmentation were observed in cats at doses of 125 mg/kg/day (multiple of 0.8 times the maximum human dose based on body surface area). Studies suggest that the underlying mechanism may be related to a transient retinal ischemia at high exposures, linked to the known sympathomimetic effect of high plasma exposures of tranexamic acid.
How Supplied/Storage and Handling
Medication Guide
17 PATIENT COUNSELING INFORMATION
Thromboembolic Risk
Inform patients that CYKLOKAPRON may increase the risk of venous and arterial thrombosis or thromboembolism and to contact their healthcare provider for any signs or symptoms suggestive of thromboembolism.
Advise patients using hormonal contraception that combined use with CYKLOKAPRON may increase the risk for thromboembolic adverse reactions and to use effective alternative (nonhormonal) contraception during therapy with CYKLOKAPRON [see Warnings and Precautions (5.1), Drug Interactions (7.1), Use in Specific Populations (8.3)].
Seizures
Inform patients that CYKLOKAPRON may cause seizures and to contact their healthcare provider for any signs or symptoms suggestive of seizures [see Warnings and Precautions (5.3)].
Hypersensitivity Reactions
Inform patients that CYKLOKAPRON may cause hypersensitivity reactions and to contact their healthcare provider for any signs or symptoms of hypersensitivity reactions [see Warnings and Precautions (5.4)].
Visual Disturbances
Inform patients that CYKLOKAPRON can cause visual disturbance and that they should report any eye symptoms or change in their vision to their healthcare provider and to follow-up with an ophthalmologist for a complete ophthalmologic evaluation, including dilated retinal examination of the retina [see Warnings and Precautions (5.5)].
Risk of Driving and Operating Machinery
Inform patients that CYKLOKAPRON may cause dizziness, and that the patient should be cautioned about driving, operating machinery, or performing hazardous tasks while taking CYKLOKAPRON [see Warnings and Precautions (5.6)].
Other
This product's labeling may have been updated. For the most recent prescribing information, please visit www.pfizer.com.

LAB-0258-17.0
17 PATIENT COUNSELING INFORMATION
17 PATIENT COUNSELING INFORMATION
Thromboembolic Risk
Inform patients that CYKLOKAPRON may increase the risk of venous and arterial thrombosis or thromboembolism and to contact their healthcare provider for any signs or symptoms suggestive of thromboembolism.
Advise patients using hormonal contraception that combined use with CYKLOKAPRON may increase the risk for thromboembolic adverse reactions and to use effective alternative (nonhormonal) contraception during therapy with CYKLOKAPRON [see Warnings and Precautions (5.1), Drug Interactions (7.1), Use in Specific Populations (8.3)].
Seizures
Inform patients that CYKLOKAPRON may cause seizures and to contact their healthcare provider for any signs or symptoms suggestive of seizures [see Warnings and Precautions (5.3)].
Hypersensitivity Reactions
Inform patients that CYKLOKAPRON may cause hypersensitivity reactions and to contact their healthcare provider for any signs or symptoms of hypersensitivity reactions [see Warnings and Precautions (5.4)].
Visual Disturbances
Inform patients that CYKLOKAPRON can cause visual disturbance and that they should report any eye symptoms or change in their vision to their healthcare provider and to follow-up with an ophthalmologist for a complete ophthalmologic evaluation, including dilated retinal examination of the retina [see Warnings and Precautions (5.5)].
Risk of Driving and Operating Machinery
Inform patients that CYKLOKAPRON may cause dizziness, and that the patient should be cautioned about driving, operating machinery, or performing hazardous tasks while taking CYKLOKAPRON [see Warnings and Precautions (5.6)].
Full Patient Information
Full Patient Information
17 PATIENT COUNSELING INFORMATION
Thromboembolic Risk
Inform patients that CYKLOKAPRON may increase the risk of venous and arterial thrombosis or thromboembolism and to contact their healthcare provider for any signs or symptoms suggestive of thromboembolism.
Advise patients using hormonal contraception that combined use with CYKLOKAPRON may increase the risk for thromboembolic adverse reactions and to use effective alternative (nonhormonal) contraception during therapy with CYKLOKAPRON [see Warnings and Precautions (5.1), Drug Interactions (7.1), Use in Specific Populations (8.3)].
Seizures
Inform patients that CYKLOKAPRON may cause seizures and to contact their healthcare provider for any signs or symptoms suggestive of seizures [see Warnings and Precautions (5.3)].
Hypersensitivity Reactions
Inform patients that CYKLOKAPRON may cause hypersensitivity reactions and to contact their healthcare provider for any signs or symptoms of hypersensitivity reactions [see Warnings and Precautions (5.4)].
Visual Disturbances
Inform patients that CYKLOKAPRON can cause visual disturbance and that they should report any eye symptoms or change in their vision to their healthcare provider and to follow-up with an ophthalmologist for a complete ophthalmologic evaluation, including dilated retinal examination of the retina [see Warnings and Precautions (5.5)].
Risk of Driving and Operating Machinery
Inform patients that CYKLOKAPRON may cause dizziness, and that the patient should be cautioned about driving, operating machinery, or performing hazardous tasks while taking CYKLOKAPRON [see Warnings and Precautions (5.6)].
Resources
Didn’t find what you were looking for?
Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5Pm ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this product, click the link below to submit your information: Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer product, please call Pfizer Medical Information at (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800)-332-1088.