BRAFTOVI® Clinical Studies
(encorafenib)
14 CLINICAL STUDIES
14.1 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
BRAFTOVI in combination with binimetinib was evaluated in a randomized, active-controlled, open-label, multicenter trial (COLUMBUS; NCT01909453). Eligible patients were required to have BRAF V600E or V600K mutation-positive unresectable or metastatic melanoma, as detected using the bioMerieux THxID™BRAF assay. Patients were permitted to have received immunotherapy in the adjuvant setting and one prior line of immunotherapy for unresectable locally advanced or metastatic disease. Prior use of BRAF inhibitors or MEK inhibitors was prohibited. Randomization was stratified by American Joint Committee on Cancer (AJCC) Stage (IIIB, IIIC, IVM1a or IVM1b, versus IVM1c), Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1), and prior immunotherapy for unresectable or metastatic disease (yes versus no).
Patients were randomized (1:1:1) to receive BRAFTOVI 450 mg once daily in combination with binimetinib 45 mg twice daily (BRAFTOVI in combination with binimetinib), BRAFTOVI 300 mg once daily, or vemurafenib 960 mg twice daily. Treatment continued until disease progression or unacceptable toxicity. Only the results of the approved dosing (BRAFTOVI 450 mg in combination with binimetinib 45 mg) are described below.
The major efficacy outcome measure was progression-free survival (PFS), as assessed by a blinded independent central review, to compare BRAFTOVI in combination with binimetinib with vemurafenib. Additional efficacy outcome measures included overall survival (OS), as well as objective response rate (ORR) and duration of response (DoR) which were assessed by central review.
A total of 577 patients were randomized, 192 to the BRAFTOVI in combination with binimetinib arm, 194 to the BRAFTOVI arm, and 191 to the vemurafenib arm. Of the 383 patients randomized to either the BRAFTOVI in combination with binimetinib or the vemurafenib arms, the median age was 56 years (20 to 89 years), 59% were male, 91% were White, and 72% had baseline ECOG performance status of 0. Ninety-five percent (95%) had metastatic disease, 65% were Stage IVM1c, and 4% received prior CTLA-4, PD-1, or PD-L1 directed antibodies. Twenty-eight percent (28%) had elevated baseline serum lactate dehydrogenase (LDH), 45% had ≥3 organs with tumor involvement at baseline, and 3% had brain metastases. Based on centralized testing, 100% of patients' tumors tested positive for BRAF mutations; BRAF V600E (88%), BRAF V600K (11%), or both (<1%).
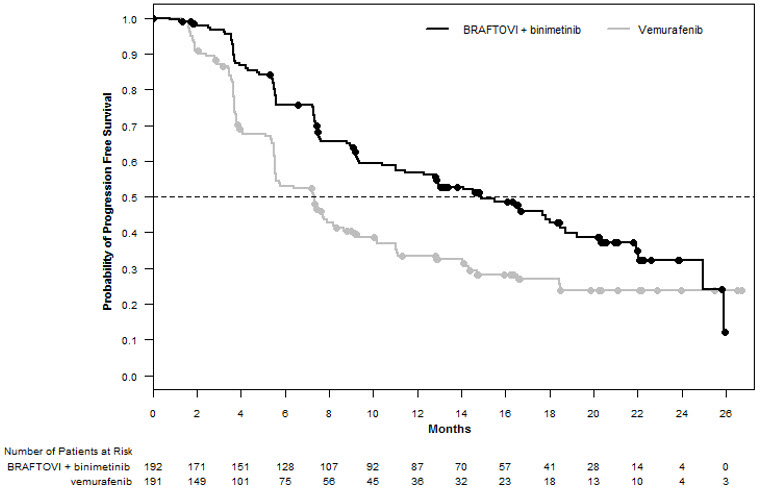
BRAFTOVI in combination with binimetinib demonstrated a statistically significant improvement in PFS compared to vemurafenib. Efficacy results are summarized in Table 11 and Figure 1.
| BRAFTOVI with binimetinib N=192 | Vemurafenib N=191 | |
|---|---|---|
| CI = Confidence interval; CR = Complete response; DoR = Duration of response; HR = Hazard ratio; NE = Not estimable; ORR = Overall response rate; OS = Overall survival; PFS = Progression-free survival; PR = Partial response. | ||
| ||
Progression-Free Survival | ||
Number of events (%) | 98 (51) | 106 (55) |
Progressive disease | 88 (46) | 104 (54) |
Death | 10 (5) | 2 (1) |
Median PFS, months (95% CI) | 14.9 (11, 18.5) | 7.3 (5.6, 8.2) |
HR (95% CI)* | 0.54 (0.41, 0.71) | |
P-value† | <0.0001 | |
Overall Survival‡ | ||
Number of events (%) | 105 (55) | 127 (67) |
Median OS, months (95% CI) | 33.6 (24.4, 39.2) | 16.9 (14.0, 24.5) |
HR (95% CI)* | 0.61 (0.47, 0.79) | |
Overall Response Rate | ||
ORR (95% CI) | 63% (56%, 70%) | 40% (33%, 48%) |
CR | 8% | 6% |
PR | 55% | 35% |
Duration of Response | ||
Median DoR, months (95% CI) | 16.6 (12.2, 20.4) | 12.3 (6.9, 16.9) |
Figure 1: Kaplan-Meier Curves for Progression-Free Survival in COLUMBUS
14.2 BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
BRAFTOVI in combination with cetuximab was evaluated in a randomized, active-controlled, open-label, multicenter trial (BEACON CRC; NCT02928224). Eligible patients were required to have BRAF V600E mutation-positive metastatic colorectal cancer (CRC), as detected using the Qiagen therascreen BRAF V600E RGQ polymerase chain reaction (PCR) Kit, with disease progression after 1 or 2 prior regimens. Other key eligibility criteria included absence of prior treatment with a RAF, MEK, or EGFR inhibitor, eligibility to receive cetuximab per local labeling with respect to tumor RAS status, and ECOG performance status (PS) 0–1. Randomization was stratified by Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1), prior use of irinotecan (yes versus no), and cetuximab product used (US-licensed versus EU-approved).
Patients were randomized 1:1:1 to one of the following treatment arms:
- •
- BRAFTOVI 300 mg orally once daily in combination with cetuximab (BRAFTOVI/cetuximab arm)
- •
- BRAFTOVI 300 mg orally once daily in combination with binimetinib and cetuximab
- •
- Irinotecan with cetuximab or FOLFIRI with cetuximab (control arm)
The dosage of cetuximab in all patients was 400 mg/m2 intravenously for the first dose followed by 250 mg/m2 weekly.
Patients in the control arm received cetuximab with either irinotecan 180 mg/m2 intravenously on Days 1 and 15 of each 28-day cycle or FOLFIRI intravenously (irinotecan 180 mg/m2 on Days 1 and 15; folinic acid 400 mg/m2 on Days 1 and 15; then fluorouracil 400 mg/m2 bolus on Days 1 and 15 followed by fluorouracil 2400 mg/m2/day by continuous infusion over 2 days).
Treatment continued until disease progression or unacceptable toxicity. Only the results of the approved regimen (BRAFTOVI in combination with cetuximab) are described below.
The major efficacy outcome measure was overall survival (OS). Additional efficacy outcome measures included progression-free survival (PFS), overall response rate (ORR), and duration of response (DoR) as assessed by blinded independent central review (BICR). OS and PFS were assessed in all randomized patients. ORR and DoR were assessed in the subset of the first 220 patients included in the randomized portion of the BRAFTOVI/cetuximab and control arm of the study.
A total of 220 patients were randomized to the BRAFTOVI/cetuximab arm and 221 to the control arm. Of these 441 patients, the median age was 61 years; 53% were female; 80% were White and 15% were Asian. Fifty percent (50%) had baseline ECOG performance status of 0; 66% received 1 prior therapy and 34% received 2; 93% received prior oxaliplatin and 52% received prior irinotecan.
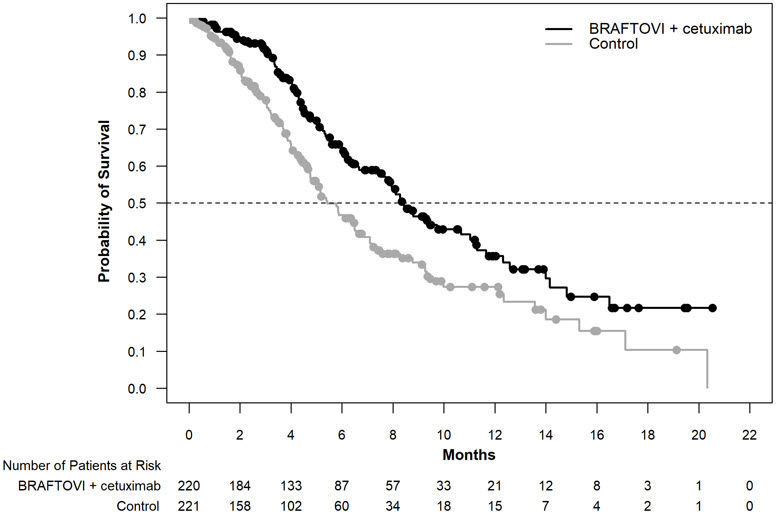
BRAFTOVI in combination with cetuximab demonstrated a statistically significant improvement in OS, ORR, and PFS compared to the active comparator. Efficacy results are summarized in Table 12 and Figure 2.
| BRAFTOVI with cetuximab N = 220 | Irinotecan with cetuximab or FOLFIRI with cetuximab N = 221 | |
|---|---|---|
| CI = Confidence interval; CR = Complete response; DoR = Duration of response; HR = Hazard ratio; NR = Not reached; ORR = Overall response rate; OS = Overall survival; PFS = Progression-free survival; PR = Partial response. | ||
| ||
Overall Survival | ||
Number of Events (%) | 93 (42) | 114 (52) |
Median OS, months (95% CI) | 8.4 (7.5, 11.0) | 5.4 (4.8, 6.6) |
0.60 (0.45, 0.79) | ||
0.0003 | ||
Overall Response Rate (per BICR) | ||
ORR (95% CI)§ | 20% (13%, 29%) | 2% (0%, 7%) |
CR | 5% | 0% |
PR | 15% | 2% |
<0.0001 | ||
Median DoR, months (95% CI) | 6.1 (4.1, 8.3) | NR (2.6, NR) |
Progression Free Survival (per BICR) | ||
Number of events (%) | 133 (60) | 128 (58) |
Progressive disease | 110 (50) | 101 (46) |
Death | 23 (10) | 27 (12) |
Median PFS, months (95% CI) | 4.2 (3.7, 5.4) | 1.5 (1.4, 1.7) |
0.40 (0.31, 0.52) | ||
< 0.0001 | ||
Figure 2: Kaplan-Meier Curves for Overall Survival in BEACON CRC
14.3 BRAF V600E Mutation-Positive Metastatic Non-Small Cell Lung Cancer
BRAFTOVI in combination with binimetinib was evaluated in an open-label, multicenter, single-arm study in patients with BRAF V600E mutation-positive metastatic non-small cell lung cancer (NSCLC) (PHAROS; NCT03915951). Eligible patients had a diagnosis of histologically-confirmed metastatic NSCLC with BRAF V600E mutation that was treatment-naïve or had been previously treated with 1 prior line of systemic therapy in the metastatic setting (platinum-based chemotherapy and/or anti-PD-1/PD-L1 therapies), age 18 years or older, Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1, and measurable disease as defined by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Prior use of BRAF inhibitors or MEK inhibitors was not allowed.
Patients received BRAFTOVI 450 mg once daily and binimetinib 45 mg orally twice daily until disease progression or unacceptable toxicity. The major efficacy outcome measures were objective response rate (ORR) per RECIST v1.1 and duration of response (DoR) as assessed by independent review committee (IRC).
In the efficacy population, BRAF V600E mutation status was determined by prospective local testing using tumor tissue (78%) or blood (22%) specimens. Of the 98 patients with BRAF V600E mutation, 6 patients were enrolled into the trial based on testing of their tumor tissue specimens with the FoundationOne CDx tissue test. Of the remaining 92 patients enrolled based on local testing, 68 patients had their tumor tissue specimens retrospectively confirmed as having BRAF V600E positive status by the FoundationOne CDx tissue test. The remaining patients had either BRAF V600E negative status (n=5) or had unevaluable results (n=19) by the FoundationOne CDx tissue test. In addition, plasma samples from 81 out of 98 patients were retrospectively tested using the FoundationOne Liquid CDx assay. Of the 81 patients, 48 were confirmed positive for BRAF V600E, while 33 patients were BRAF V600E mutation negative by FoundationOne Liquid CDx assay. The remaining 17 samples had unevaluable results with FoundationOne Liquid CDx assay.
The efficacy population included 59 treatment-naïve patients and 39 previously-treated patients. Among these 98 patients, the median age was 70 years (range: 47 to 86); 53% female; 88% White, 7% Asian, 3% Black or African American, and 1% American Indian or Alaska Native; 99% were not Hispanic or Latino; 13% were current smokers and 57% were former smokers; 73% had ECOG PS of 1; and 97% had adenocarcinoma. All patients had metastatic disease, and 8% had brain metastases at baseline.
Efficacy results for patients with BRAF V600E mutation-positive metastatic NSCLC are summarized in Table 13.
| CI = Confidence interval; CR = Complete response; DoR = Duration of response; N = Number of patients; NE = Not estimable; ORR = Objective response rate; PR = Partial response. | ||
| ||
BRAFTOVI with binimetinib | ||
Efficacy Parameter | Treatment naïve (N=59) | Previously treated (N=39) |
Objective Response Rate* | ||
ORR (95% CI) | 75% (62, 85) | 46% (30, 63) |
CR | 15% | 10% |
PR | 59% | 36% |
Duration of Response* | N=44 | N=18 |
Median DoR, months (95% CI) | NE (23.1, NE) | 16.7 (7.4, NE) |
% with DoR ≥6 months | 75% | 67% |
% with DoR ≥12 months | 59% | 33% |
Find BRAFTOVI® medical information:
Find BRAFTOVI® medical information:
BRAFTOVI® Quick Finder
Health Professional Information
Clinical Studies
14 CLINICAL STUDIES
14.1 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
BRAFTOVI in combination with binimetinib was evaluated in a randomized, active-controlled, open-label, multicenter trial (COLUMBUS; NCT01909453). Eligible patients were required to have BRAF V600E or V600K mutation-positive unresectable or metastatic melanoma, as detected using the bioMerieux THxID™BRAF assay. Patients were permitted to have received immunotherapy in the adjuvant setting and one prior line of immunotherapy for unresectable locally advanced or metastatic disease. Prior use of BRAF inhibitors or MEK inhibitors was prohibited. Randomization was stratified by American Joint Committee on Cancer (AJCC) Stage (IIIB, IIIC, IVM1a or IVM1b, versus IVM1c), Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1), and prior immunotherapy for unresectable or metastatic disease (yes versus no).
Patients were randomized (1:1:1) to receive BRAFTOVI 450 mg once daily in combination with binimetinib 45 mg twice daily (BRAFTOVI in combination with binimetinib), BRAFTOVI 300 mg once daily, or vemurafenib 960 mg twice daily. Treatment continued until disease progression or unacceptable toxicity. Only the results of the approved dosing (BRAFTOVI 450 mg in combination with binimetinib 45 mg) are described below.
The major efficacy outcome measure was progression-free survival (PFS), as assessed by a blinded independent central review, to compare BRAFTOVI in combination with binimetinib with vemurafenib. Additional efficacy outcome measures included overall survival (OS), as well as objective response rate (ORR) and duration of response (DoR) which were assessed by central review.
A total of 577 patients were randomized, 192 to the BRAFTOVI in combination with binimetinib arm, 194 to the BRAFTOVI arm, and 191 to the vemurafenib arm. Of the 383 patients randomized to either the BRAFTOVI in combination with binimetinib or the vemurafenib arms, the median age was 56 years (20 to 89 years), 59% were male, 91% were White, and 72% had baseline ECOG performance status of 0. Ninety-five percent (95%) had metastatic disease, 65% were Stage IVM1c, and 4% received prior CTLA-4, PD-1, or PD-L1 directed antibodies. Twenty-eight percent (28%) had elevated baseline serum lactate dehydrogenase (LDH), 45% had ≥3 organs with tumor involvement at baseline, and 3% had brain metastases. Based on centralized testing, 100% of patients' tumors tested positive for BRAF mutations; BRAF V600E (88%), BRAF V600K (11%), or both (<1%).
BRAFTOVI in combination with binimetinib demonstrated a statistically significant improvement in PFS compared to vemurafenib. Efficacy results are summarized in Table 11 and Figure 1.
| BRAFTOVI with binimetinib N=192 | Vemurafenib N=191 | |
|---|---|---|
| CI = Confidence interval; CR = Complete response; DoR = Duration of response; HR = Hazard ratio; NE = Not estimable; ORR = Overall response rate; OS = Overall survival; PFS = Progression-free survival; PR = Partial response. | ||
| ||
Progression-Free Survival | ||
Number of events (%) | 98 (51) | 106 (55) |
Progressive disease | 88 (46) | 104 (54) |
Death | 10 (5) | 2 (1) |
Median PFS, months (95% CI) | 14.9 (11, 18.5) | 7.3 (5.6, 8.2) |
HR (95% CI)* | 0.54 (0.41, 0.71) | |
P-value† | <0.0001 | |
Overall Survival‡ | ||
Number of events (%) | 105 (55) | 127 (67) |
Median OS, months (95% CI) | 33.6 (24.4, 39.2) | 16.9 (14.0, 24.5) |
HR (95% CI)* | 0.61 (0.47, 0.79) | |
Overall Response Rate | ||
ORR (95% CI) | 63% (56%, 70%) | 40% (33%, 48%) |
CR | 8% | 6% |
PR | 55% | 35% |
Duration of Response | ||
Median DoR, months (95% CI) | 16.6 (12.2, 20.4) | 12.3 (6.9, 16.9) |
Figure 1: Kaplan-Meier Curves for Progression-Free Survival in COLUMBUS
14.2 BRAF V600E Mutation-Positive Metastatic Colorectal Cancer (CRC)
BRAFTOVI in combination with cetuximab was evaluated in a randomized, active-controlled, open-label, multicenter trial (BEACON CRC; NCT02928224). Eligible patients were required to have BRAF V600E mutation-positive metastatic colorectal cancer (CRC), as detected using the Qiagen therascreen BRAF V600E RGQ polymerase chain reaction (PCR) Kit, with disease progression after 1 or 2 prior regimens. Other key eligibility criteria included absence of prior treatment with a RAF, MEK, or EGFR inhibitor, eligibility to receive cetuximab per local labeling with respect to tumor RAS status, and ECOG performance status (PS) 0–1. Randomization was stratified by Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1), prior use of irinotecan (yes versus no), and cetuximab product used (US-licensed versus EU-approved).
Patients were randomized 1:1:1 to one of the following treatment arms:
- •
- BRAFTOVI 300 mg orally once daily in combination with cetuximab (BRAFTOVI/cetuximab arm)
- •
- BRAFTOVI 300 mg orally once daily in combination with binimetinib and cetuximab
- •
- Irinotecan with cetuximab or FOLFIRI with cetuximab (control arm)
The dosage of cetuximab in all patients was 400 mg/m2 intravenously for the first dose followed by 250 mg/m2 weekly.
Patients in the control arm received cetuximab with either irinotecan 180 mg/m2 intravenously on Days 1 and 15 of each 28-day cycle or FOLFIRI intravenously (irinotecan 180 mg/m2 on Days 1 and 15; folinic acid 400 mg/m2 on Days 1 and 15; then fluorouracil 400 mg/m2 bolus on Days 1 and 15 followed by fluorouracil 2400 mg/m2/day by continuous infusion over 2 days).
Treatment continued until disease progression or unacceptable toxicity. Only the results of the approved regimen (BRAFTOVI in combination with cetuximab) are described below.
The major efficacy outcome measure was overall survival (OS). Additional efficacy outcome measures included progression-free survival (PFS), overall response rate (ORR), and duration of response (DoR) as assessed by blinded independent central review (BICR). OS and PFS were assessed in all randomized patients. ORR and DoR were assessed in the subset of the first 220 patients included in the randomized portion of the BRAFTOVI/cetuximab and control arm of the study.
A total of 220 patients were randomized to the BRAFTOVI/cetuximab arm and 221 to the control arm. Of these 441 patients, the median age was 61 years; 53% were female; 80% were White and 15% were Asian. Fifty percent (50%) had baseline ECOG performance status of 0; 66% received 1 prior therapy and 34% received 2; 93% received prior oxaliplatin and 52% received prior irinotecan.
BRAFTOVI in combination with cetuximab demonstrated a statistically significant improvement in OS, ORR, and PFS compared to the active comparator. Efficacy results are summarized in Table 12 and Figure 2.
| BRAFTOVI with cetuximab N = 220 | Irinotecan with cetuximab or FOLFIRI with cetuximab N = 221 | |
|---|---|---|
| CI = Confidence interval; CR = Complete response; DoR = Duration of response; HR = Hazard ratio; NR = Not reached; ORR = Overall response rate; OS = Overall survival; PFS = Progression-free survival; PR = Partial response. | ||
| ||
Overall Survival | ||
Number of Events (%) | 93 (42) | 114 (52) |
Median OS, months (95% CI) | 8.4 (7.5, 11.0) | 5.4 (4.8, 6.6) |
0.60 (0.45, 0.79) | ||
0.0003 | ||
Overall Response Rate (per BICR) | ||
ORR (95% CI)§ | 20% (13%, 29%) | 2% (0%, 7%) |
CR | 5% | 0% |
PR | 15% | 2% |
<0.0001 | ||
Median DoR, months (95% CI) | 6.1 (4.1, 8.3) | NR (2.6, NR) |
Progression Free Survival (per BICR) | ||
Number of events (%) | 133 (60) | 128 (58) |
Progressive disease | 110 (50) | 101 (46) |
Death | 23 (10) | 27 (12) |
Median PFS, months (95% CI) | 4.2 (3.7, 5.4) | 1.5 (1.4, 1.7) |
0.40 (0.31, 0.52) | ||
< 0.0001 | ||
Figure 2: Kaplan-Meier Curves for Overall Survival in BEACON CRC
14.3 BRAF V600E Mutation-Positive Metastatic Non-Small Cell Lung Cancer
BRAFTOVI in combination with binimetinib was evaluated in an open-label, multicenter, single-arm study in patients with BRAF V600E mutation-positive metastatic non-small cell lung cancer (NSCLC) (PHAROS; NCT03915951). Eligible patients had a diagnosis of histologically-confirmed metastatic NSCLC with BRAF V600E mutation that was treatment-naïve or had been previously treated with 1 prior line of systemic therapy in the metastatic setting (platinum-based chemotherapy and/or anti-PD-1/PD-L1 therapies), age 18 years or older, Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1, and measurable disease as defined by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Prior use of BRAF inhibitors or MEK inhibitors was not allowed.
Patients received BRAFTOVI 450 mg once daily and binimetinib 45 mg orally twice daily until disease progression or unacceptable toxicity. The major efficacy outcome measures were objective response rate (ORR) per RECIST v1.1 and duration of response (DoR) as assessed by independent review committee (IRC).
In the efficacy population, BRAF V600E mutation status was determined by prospective local testing using tumor tissue (78%) or blood (22%) specimens. Of the 98 patients with BRAF V600E mutation, 6 patients were enrolled into the trial based on testing of their tumor tissue specimens with the FoundationOne CDx tissue test. Of the remaining 92 patients enrolled based on local testing, 68 patients had their tumor tissue specimens retrospectively confirmed as having BRAF V600E positive status by the FoundationOne CDx tissue test. The remaining patients had either BRAF V600E negative status (n=5) or had unevaluable results (n=19) by the FoundationOne CDx tissue test. In addition, plasma samples from 81 out of 98 patients were retrospectively tested using the FoundationOne Liquid CDx assay. Of the 81 patients, 48 were confirmed positive for BRAF V600E, while 33 patients were BRAF V600E mutation negative by FoundationOne Liquid CDx assay. The remaining 17 samples had unevaluable results with FoundationOne Liquid CDx assay.
The efficacy population included 59 treatment-naïve patients and 39 previously-treated patients. Among these 98 patients, the median age was 70 years (range: 47 to 86); 53% female; 88% White, 7% Asian, 3% Black or African American, and 1% American Indian or Alaska Native; 99% were not Hispanic or Latino; 13% were current smokers and 57% were former smokers; 73% had ECOG PS of 1; and 97% had adenocarcinoma. All patients had metastatic disease, and 8% had brain metastases at baseline.
Efficacy results for patients with BRAF V600E mutation-positive metastatic NSCLC are summarized in Table 13.
| CI = Confidence interval; CR = Complete response; DoR = Duration of response; N = Number of patients; NE = Not estimable; ORR = Objective response rate; PR = Partial response. | ||
| ||
BRAFTOVI with binimetinib | ||
Efficacy Parameter | Treatment naïve (N=59) | Previously treated (N=39) |
Objective Response Rate* | ||
ORR (95% CI) | 75% (62, 85) | 46% (30, 63) |
CR | 15% | 10% |
PR | 59% | 36% |
Duration of Response* | N=44 | N=18 |
Median DoR, months (95% CI) | NE (23.1, NE) | 16.7 (7.4, NE) |
% with DoR ≥6 months | 75% | 67% |
% with DoR ≥12 months | 59% | 33% |
Health Professional Information
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5Pm ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.