ELLENCE® Warnings and Precautions
(epirubicin HCl)
5 WARNINGS AND PRECAUTIONS
5.1 Cardiac Toxicity
ELLENCE and other anthracycline drugs can result in either early (or acute) or late (delayed) cardiac toxicity.
The principal manifestations of early cardiac toxicity are sinus tachycardia and/or electrocardiogram (ECG) abnormalities such as non-specific ST-T wave changes. However, tachycardia (including premature ventricular contractions and ventricular tachycardia), bradycardia, as well as atrioventricular and bundle-branch block have been reported. Early cardiac toxicity does not usually predict the subsequent occurrence of delayed cardiotoxicity and generally should not be considered a reason for suspending treatment with ELLENCE.
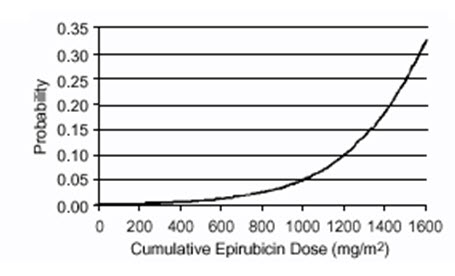
Delayed cardiac toxicity is manifested by reduced left ventricular ejection fraction (LVEF) and/or signs and symptoms of congestive heart failure (CHF). If it occurs, late cardiotoxicity usually develops late during therapy with ELLENCE or within 2 to 3 months after completion of treatment, but there are reports of it occurring several months to years after treatment termination. In a retrospective survey, including 9144 patients, mostly with solid tumors in advanced stages, the probability of developing CHF increased with increasing cumulative doses of ELLENCE (Figure 1). In another retrospective survey of 469 ELLENCE-treated patients with metastatic or early breast cancer, the reported risk of CHF was comparable to that observed in the larger study of over 9000 patients.
Given the risk of cardiac toxicity, cumulative doses of 900 mg/m2 ELLENCE should generally be avoided.
Figure 1. Risk of CHF in 9144 Patients Treated with ELLENCE
Prior history of cardiovascular disease, prior or concomitant radiotherapy to the mediastinal/pericardial area, previous therapy with other anthracyclines or anthracenediones, and concomitant use of other cardiotoxic drugs, increase the risk of developing late cardiac toxicity. Avoid administration of ELLENCE in combination with other cardiotoxic drugs. Although not formally tested, it is probable that the toxicity of ELLENCE and other anthracyclines or anthracenediones is additive. Cardiac toxicity with ELLENCE may occur at lower cumulative doses whether or not cardiac risk factors are present. Patients receiving ELLENCE after stopping treatment with other cardiotoxic drugs, especially those with long half-lives such as trastuzumab, may be at increased risk of developing cardiotoxicity [see Dosage and Administration (2) and Drug Interaction (7.1)].
Perform a baseline ECG and evaluation of LVEF prior to initiating treatment with ELLENCE. Monitor LVEF during the course of treatment and consider discontinuation of ELLENCE if LVEF decrease and/or signs or symptoms of CHF develop. Closely monitor patients with other risk factors for cardiac toxicity, particularly prior administration of anthracycline or anthracenedione.
5.2 Secondary Malignancies
The risk of developing secondary acute myelogenous leukemia and myelodysplastic syndrome (MDS), is increased following treatment with ELLENCE and other anthracyclines. Cumulative risk of secondary acute myelogenous leukemia or myelodysplastic syndrome (AML/MDS) of about 0.27% at 3 years, 0.46% at 5 years, and 0.55% at 8 years. These leukemias generally occur within 1 to 3 years of treatment [see Adverse Reactions (6.1)].
5.3 Extravasation and Tissue Necrosis
Extravasation of ELLENCE can result in severe local tissue injury manifesting as blistering, ulceration, and necrosis requiring wide excision of the affected area and skin grafting. Extravasation should be considered if a patient experiences a burning or stinging sensation or shows other evidence indicating peri-venous infiltration or extravasation; however, extravasation may be present in patients who do not experience a stinging or burning sensation or when blood return is present on aspiration of the infusion needle.
Venous sclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Administer ELLENCE slowly into the tubing of a freely running intravenous infusion. Patients receiving initial therapy at the recommended starting doses of 100–120 mg/m2 should have ELLENCE infused over 15–20 minutes. For patients who require lower ELLENCE starting doses due to organ dysfunction or who require modification of ELLENCE doses during therapy, the ELLENCE infusion time may be proportionally decreased, but should not be less than 3 minutes [see Dosage and Administration (2.3)]. If possible, avoid veins over joints or in extremities with compromised venous or lymphatic drainage. Facial flushing, as well as local erythematous streaking along the vein, may be indicative of excessively rapid administration. It may precede local phlebitis or thrombophlebitis.
Immediately terminate infusion and restart in another vein if a burning or stinging sensation indicates perivenous infiltration. Perivenous infiltration may occur without causing pain. If extravasation is suspected, immediately discontinue the intravenous injection or continuous intravenous infusion [see Dosage and Administration (2.3)]. Apply ice to the site intermittently for 15 minutes, 4 times a day for 3 days. If appropriate, administer dexrazoxane at the site of extravasation as soon as possible and within the first 6 hours after extravasation.
5.4 Severe Myelosuppression
ELLENCE can cause severe myelosuppression [see Adverse Reactions (6.1). Obtain complete blood counts prior to each treatment and carefully monitor patients during treatment for possible clinical complications due to myelosuppression. Delay the next dose of ELLENCE if severe myelosuppression has not improved. Consider dose reduction for patients with prolonged myelosuppression based on the severity of reaction [see Dosage and Administration (2.3)].
5.5 Use in Patients with Hepatic Impairment
The major route of elimination of epirubicin is the hepatobiliary system [see Clinical Pharmacology (12.3)]. Evaluate serum total bilirubin and AST levels before and during treatment with ELLENCE. Patients with elevated bilirubin or AST may experience slower clearance of drug with an increase in overall toxicity. Lower doses are recommended in these patients [see Dosage and Administration (2.2)]. Patients with severe hepatic impairment have not been evaluated; therefore, do not use ELLENCE in this patient population [see Contraindications (4)].
5.6 Use in Patients with Renal Impairment
Assess serum creatinine before and during therapy. Dosage adjustment is necessary in patients with serum creatinine >5 mg/dL [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)]. Patients undergoing dialysis have not been studied.
5.7 Tumor-Lysis Syndrome
ELLENCE can induce tumor lysis syndrome in patients with rapidly growing tumors. Evaluate blood uric acid levels, potassium, calcium, phosphate, and creatinine after initial treatment. Consider hydration, urine alkalinization, and prophylaxis with allopurinol to minimize hyperuricemia and potential complications of tumor lysis syndrome.
5.8 Immunosuppressant Effects/Increased Susceptibility to Infections
Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including epirubicin, may result in serious or fatal infections. Avoid vaccination with a live vaccine in patients receiving ELLENCE. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
5.9 Thrombophlebitis and Thromboembolic Events
Thrombophlebitis and thromboembolic events, including pulmonary embolism (in some cases fatal) have been reported with the use of ELLENCE.
5.10 Potentiation of Radiation Toxicity and Radiation Recall
ELLENCE can increase radiation-induced toxicity to the myocardium, mucosa, skin, and liver. Radiation recall, including but not limited to cutaneous and pulmonary toxicity, can occur in patients who receive ELLENCE after prior radiation therapy.
5.11 Embryo-Fetal Toxicity
Based on findings from animals and its mechanism of action, ELLENCE can cause fetal harm when administered to a pregnant woman; avoid the use of ELLENCE during the 1st trimester. Available human data do not establish the presence or absence of major birth defects and miscarriage related to the use of epirubicin during the 2nd and 3rd trimesters. In animal reproduction studies, epirubicin was embryo-fetal lethal and caused structural abnormalities in rats and rabbits at doses less than the maximum recommended human dose on a body surface area basis. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise female patients of reproductive potential to use effective contraception during treatment and for 6 months after the last dose of ELLENCE. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of ELLENCE. Advise male patients with pregnant partners to use condoms during treatment and for at least 7 days after the last dose of ELLENCE [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1), and Nonclinical Toxicology (13.1)].
Find ELLENCE® medical information:
Find ELLENCE® medical information:
ELLENCE® Quick Finder
Health Professional Information
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Cardiac Toxicity
ELLENCE and other anthracycline drugs can result in either early (or acute) or late (delayed) cardiac toxicity.
The principal manifestations of early cardiac toxicity are sinus tachycardia and/or electrocardiogram (ECG) abnormalities such as non-specific ST-T wave changes. However, tachycardia (including premature ventricular contractions and ventricular tachycardia), bradycardia, as well as atrioventricular and bundle-branch block have been reported. Early cardiac toxicity does not usually predict the subsequent occurrence of delayed cardiotoxicity and generally should not be considered a reason for suspending treatment with ELLENCE.
Delayed cardiac toxicity is manifested by reduced left ventricular ejection fraction (LVEF) and/or signs and symptoms of congestive heart failure (CHF). If it occurs, late cardiotoxicity usually develops late during therapy with ELLENCE or within 2 to 3 months after completion of treatment, but there are reports of it occurring several months to years after treatment termination. In a retrospective survey, including 9144 patients, mostly with solid tumors in advanced stages, the probability of developing CHF increased with increasing cumulative doses of ELLENCE (Figure 1). In another retrospective survey of 469 ELLENCE-treated patients with metastatic or early breast cancer, the reported risk of CHF was comparable to that observed in the larger study of over 9000 patients.
Given the risk of cardiac toxicity, cumulative doses of 900 mg/m2 ELLENCE should generally be avoided.
Figure 1. Risk of CHF in 9144 Patients Treated with ELLENCE
Prior history of cardiovascular disease, prior or concomitant radiotherapy to the mediastinal/pericardial area, previous therapy with other anthracyclines or anthracenediones, and concomitant use of other cardiotoxic drugs, increase the risk of developing late cardiac toxicity. Avoid administration of ELLENCE in combination with other cardiotoxic drugs. Although not formally tested, it is probable that the toxicity of ELLENCE and other anthracyclines or anthracenediones is additive. Cardiac toxicity with ELLENCE may occur at lower cumulative doses whether or not cardiac risk factors are present. Patients receiving ELLENCE after stopping treatment with other cardiotoxic drugs, especially those with long half-lives such as trastuzumab, may be at increased risk of developing cardiotoxicity [see Dosage and Administration (2) and Drug Interaction (7.1)].
Perform a baseline ECG and evaluation of LVEF prior to initiating treatment with ELLENCE. Monitor LVEF during the course of treatment and consider discontinuation of ELLENCE if LVEF decrease and/or signs or symptoms of CHF develop. Closely monitor patients with other risk factors for cardiac toxicity, particularly prior administration of anthracycline or anthracenedione.
5.2 Secondary Malignancies
The risk of developing secondary acute myelogenous leukemia and myelodysplastic syndrome (MDS), is increased following treatment with ELLENCE and other anthracyclines. Cumulative risk of secondary acute myelogenous leukemia or myelodysplastic syndrome (AML/MDS) of about 0.27% at 3 years, 0.46% at 5 years, and 0.55% at 8 years. These leukemias generally occur within 1 to 3 years of treatment [see Adverse Reactions (6.1)].
5.3 Extravasation and Tissue Necrosis
Extravasation of ELLENCE can result in severe local tissue injury manifesting as blistering, ulceration, and necrosis requiring wide excision of the affected area and skin grafting. Extravasation should be considered if a patient experiences a burning or stinging sensation or shows other evidence indicating peri-venous infiltration or extravasation; however, extravasation may be present in patients who do not experience a stinging or burning sensation or when blood return is present on aspiration of the infusion needle.
Venous sclerosis may result from an injection into a small vessel or from repeated injections into the same vein. Administer ELLENCE slowly into the tubing of a freely running intravenous infusion. Patients receiving initial therapy at the recommended starting doses of 100–120 mg/m2 should have ELLENCE infused over 15–20 minutes. For patients who require lower ELLENCE starting doses due to organ dysfunction or who require modification of ELLENCE doses during therapy, the ELLENCE infusion time may be proportionally decreased, but should not be less than 3 minutes [see Dosage and Administration (2.3)]. If possible, avoid veins over joints or in extremities with compromised venous or lymphatic drainage. Facial flushing, as well as local erythematous streaking along the vein, may be indicative of excessively rapid administration. It may precede local phlebitis or thrombophlebitis.
Immediately terminate infusion and restart in another vein if a burning or stinging sensation indicates perivenous infiltration. Perivenous infiltration may occur without causing pain. If extravasation is suspected, immediately discontinue the intravenous injection or continuous intravenous infusion [see Dosage and Administration (2.3)]. Apply ice to the site intermittently for 15 minutes, 4 times a day for 3 days. If appropriate, administer dexrazoxane at the site of extravasation as soon as possible and within the first 6 hours after extravasation.
5.4 Severe Myelosuppression
ELLENCE can cause severe myelosuppression [see Adverse Reactions (6.1). Obtain complete blood counts prior to each treatment and carefully monitor patients during treatment for possible clinical complications due to myelosuppression. Delay the next dose of ELLENCE if severe myelosuppression has not improved. Consider dose reduction for patients with prolonged myelosuppression based on the severity of reaction [see Dosage and Administration (2.3)].
5.5 Use in Patients with Hepatic Impairment
The major route of elimination of epirubicin is the hepatobiliary system [see Clinical Pharmacology (12.3)]. Evaluate serum total bilirubin and AST levels before and during treatment with ELLENCE. Patients with elevated bilirubin or AST may experience slower clearance of drug with an increase in overall toxicity. Lower doses are recommended in these patients [see Dosage and Administration (2.2)]. Patients with severe hepatic impairment have not been evaluated; therefore, do not use ELLENCE in this patient population [see Contraindications (4)].
5.6 Use in Patients with Renal Impairment
Assess serum creatinine before and during therapy. Dosage adjustment is necessary in patients with serum creatinine >5 mg/dL [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)]. Patients undergoing dialysis have not been studied.
5.7 Tumor-Lysis Syndrome
ELLENCE can induce tumor lysis syndrome in patients with rapidly growing tumors. Evaluate blood uric acid levels, potassium, calcium, phosphate, and creatinine after initial treatment. Consider hydration, urine alkalinization, and prophylaxis with allopurinol to minimize hyperuricemia and potential complications of tumor lysis syndrome.
5.8 Immunosuppressant Effects/Increased Susceptibility to Infections
Administration of live or live-attenuated vaccines in patients immunocompromised by chemotherapeutic agents including epirubicin, may result in serious or fatal infections. Avoid vaccination with a live vaccine in patients receiving ELLENCE. Killed or inactivated vaccines may be administered; however, the response to such vaccines may be diminished.
5.9 Thrombophlebitis and Thromboembolic Events
Thrombophlebitis and thromboembolic events, including pulmonary embolism (in some cases fatal) have been reported with the use of ELLENCE.
5.10 Potentiation of Radiation Toxicity and Radiation Recall
ELLENCE can increase radiation-induced toxicity to the myocardium, mucosa, skin, and liver. Radiation recall, including but not limited to cutaneous and pulmonary toxicity, can occur in patients who receive ELLENCE after prior radiation therapy.
5.11 Embryo-Fetal Toxicity
Based on findings from animals and its mechanism of action, ELLENCE can cause fetal harm when administered to a pregnant woman; avoid the use of ELLENCE during the 1st trimester. Available human data do not establish the presence or absence of major birth defects and miscarriage related to the use of epirubicin during the 2nd and 3rd trimesters. In animal reproduction studies, epirubicin was embryo-fetal lethal and caused structural abnormalities in rats and rabbits at doses less than the maximum recommended human dose on a body surface area basis. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise female patients of reproductive potential to use effective contraception during treatment and for 6 months after the last dose of ELLENCE. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of ELLENCE. Advise male patients with pregnant partners to use condoms during treatment and for at least 7 days after the last dose of ELLENCE [see Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1), and Nonclinical Toxicology (13.1)].
Health Professional Information
{{section_name_patient}}
{{section_body_html_patient}}
Resources
Didn’t find what you were looking for? Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
Pfizer Safety
To report an adverse event related to the Pfizer-BioNTech COVID-19 Vaccine, and you are not part of a clinical trial* for this product, click the link below to submit your information:
Pfizer Safety Reporting Site*If you are involved in a clinical trial for this product, adverse events should be reported to your coordinating study site.
If you cannot use the above website, or would like to report an adverse event related to a different Pfizer product, please call Pfizer Safety at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or call (800) 822-7967.