deferoxamine mesylate for injection, USP
Find deferoxamine mesylate for injection, USP medical information:
Find deferoxamine mesylate for injection, USP medical information:
deferoxamine mesylate for injection, USP Quick Finder
Highlights
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use DEFEROXAMINE MESYLATE safely and effectively. See full prescribing information for DEFEROXAMINE MESYLATE. DEFEROXAMINE MESYLATE for injection, for intramuscular, intravenous, or subcutaneous use Initial U.S. Approval: 1968 RECENT MAJOR CHANGES
INDICATIONS AND USAGEDeferoxamine mesylate is an iron-chelating agent indicated:
Limitations of Use Deferoxamine mesylate is not indicated for the treatment of primary hemochromatosis (since phlebotomy is the method of choice for removing excess iron in this disorder). DOSAGE AND ADMINISTRATIONAcute Iron Intoxication: (2.1)
Chronic Iron Overload: (2.2)
See Full Prescribing Information for instructions on preparation of Deferoxamine mesylate for administration. (2.3) Vitamin C (up to 200 mg) increases availability of iron for chelation and may be given as an adjuvant to iron chelation therapy. (2.4) DOSAGE FORMS AND STRENGTHSCONTRAINDICATIONSWARNINGS AND PRECAUTIONS
ADVERSE REACTIONSMost common adverse reactions are injection reactions (local and systemic), hypersensitivity reactions, infections with Yersinia and Mucormycosis, cardiovascular, gastrointestinal, hematologic, hepatic, musculoskeletal, urogenital, nervous, respiratory, ocular and hearing. (6) To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONSUSE IN SPECIFIC POPULATIONSSee 17 for PATIENT COUNSELING INFORMATION. Revised: 11/2023 |
Indications and Usage
1 INDICATIONS AND USAGE
1.1 Acute Iron Intoxication
Deferoxamine Mesylate for injection is indicated as an adjunct to standard measures for the treatment of acute iron intoxication.
Dosage and Administration
2 DOSAGE AND ADMINISTRATION
The dosage (based on body weight in mg/kg/day), rates of administration, and mode of administration for both adults and pediatric patients are individually determined and adapted during the course of therapy based on the severity of the patient's iron overload. The minimum daily dose of deferoxamine mesylate is 20 mg/kg/day for both adults and pediatric patients. The maximum daily dose is 40 mg/kg/day for pediatric patients and 60 mg/kg/day for adults.
2.1 Recommended Dosage for Treatment of Acute Iron Intoxication for Adults and Pediatric Patients
Intramuscular (IM) Administration
Use for all patients not in shock.
The initial recommended dose of Deferoxamine Mesylate for injection is 1,000 mg intramuscularly (IM) once. If needed based on the clinical response, administer subsequent doses of 500 mg every 4 hours to 12 hours. The maximum recommended daily dose is 6,000 mg in 24 hours.
Intravenous (IV) Administration
Administer Deferoxamine Mesylate for injection intravenously (IV) to patients in a state of cardiovascular collapse and then only by slow infusion. As soon as the clinical condition of the patient permits, intravenous administration should be discontinued, and the drug should be administered intramuscularly.
The initial recommended IV dose of Deferoxamine Mesylate for injection is 1,000 mg administered at an infusion rate of up to 15 mg/kg/hr. If needed based on the clinical response administer additional doses of 500 mg over 4 hours to 12 hours at a slower infusion rate of up to 125 mg/hr. The maximum recommended daily dose is 6,000 mg in 24 hours.
2.2 Recommended Dosage for Treatment of Chronic Iron Overload for Adults and Pediatric Patients
Subcutaneous Infusion Administration
The average daily dose of Deferoxamine Mesylate for injection is usually between 20 and 60 mg/kg. In general patients with serum ferritin level below 2,000 ng/mL require about 25 mg/kg/day. Patients with serum ferritin level between 2,000 and 3,000 ng/mL require about 35 mg/kg/day. Patients with higher serum ferritin may require up to 55 mg/kg/day. It is not advisable to regularly exceed an average daily dose of 50 mg/kg/day except when very intensive chelation is needed in patients who have completed growth. If ferritin levels fall below 1,000 ng/mL, the risk of Deferoxamine mesylate toxicity increases; it is important to monitor these patients particularly carefully and perhaps to consider lowering the total weekly dose. The doses specified here are the average daily doses. Since most patients use Deferoxamine mesylate less than 7 days a week, the actual dose per infusion usually differs from the average daily dose; e.g. if an average daily dose of 40 mg/kg/day is required and the patient wears the pump 5 nights a week, each infusion should contain 56 mg/kg.
Slow subcutaneous infusion using a portable, light-weight infusion pump over a period of 8 to 12 hours is regarded as effective and especially convenient for ambulatory patients, but may also be given over a 24-hour period. Deferoxamine mesylate should normally be used with the pump 5 to 7 times a week. Deferoxamine mesylate is not formulated to support subcutaneous bolus injection.
Intravenous Administration
Deferoxamine Mesylate for injection can be administered intravenously if needed in patients with intravenous access.
The recommended dose of Deferoxamine Mesylate for injection in adults is 40 mg/kg/day to 50 mg/kg/day over 8 hours to 12 hours at a rate of up to 15 mg/kg/hour for 5 days to 7 days per week. Maximum dose is 60 mg/kg/day.
The recommended dose of Deferoxamine Mesylate for injection in pediatric patients is 20 mg/kg/day to 40 mg/kg/day over 8 hours to 12 hours for 5 days to 7 days per week. The maximum recommended daily dose is 40 mg/kg/day until growth (body weight and linear growth) has ceased.
In case of missed doses, Deferoxamine Mesylate for injection may be administered prior to or following same day blood transfusion (for example, 1 gram over 4 hours on the day of transfusion); however, the contribution of this mode of administration to iron balance is limited. Deferoxamine Mesylate for injection should not be administered concurrently with the blood transfusion as this can lead to errors in interpreting side effects such as rash, anaphylaxis and hypotension.
Intramuscular Administration
If given intramuscularly, the recommended dose of Deferoxamine Mesylate for injection is 500 mg to 1,000 mg per day. The maximum recommended daily dose is 1,000 mg per day.
2.3 Preparation
Reconstitute Deferoxamine Mesylate for injection prior to administration. Deferoxamine Mesylate for injection should be further diluted for intravenous infusion. Use appropriate aseptic technique.
Reconstitute each vial of Deferoxamine Mesylate for injection with Sterile Water for Injection, USP per Table 1. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if visibly opaque particles, discoloration or foreign particles are observed. The reconstituted Deferoxamine Mesylate for injection solution is an isotonic, clear and colorless to slightly-yellowish solution. Discard unused portion.
Table 1 Preparation of Deferoxamine Mesylate for injection Prior to Administration
| |||
Vial Size | Route of Administration | Amount of Sterile Water for Injection, USP for Reconstitution | Concentration After Reconstitution |
500 mg | Intramuscular | 2 mL | 213 mg/mL |
500 mg | Intravenous* | 5 mL | 95 mg/mL† |
500 mg | Subcutaneous | 5 mL | 95 mg/mL |
2 g | Intramuscular | 8 mL | 213 mg/mL |
2 g | Intravenous | 20 mL | 95 mg/mL |
2 g | Subcutaneous | 20 mL | 95 mg/mL |
If not used immediately, store at room temperature between 20°C to 25°C (68°F to 77°F), excursions permitted between 15°C and 30°C (59°F and 86°F), for a maximum period of 24 hours. Do not refrigerate reconstituted solution.
2.4 Management of Vitamin C Deficiency
Patients with iron overload usually become vitamin C deficient, probably because iron oxidizes the vitamin. As an adjuvant to iron chelation therapy, vitamin C in doses up to 200 mg for adults may be given in divided doses, starting after an initial month of regular treatment with Deferoxamine Mesylate for injection [see Warnings and Precautions (5.7)]. Vitamin C increases availability of iron for chelation. In general, 50 mg daily suffices for pediatric patients under 10 years old and 100 mg daily for older pediatric patients. Larger doses of vitamin C fail to produce any additional increase in excretion of iron complex.
Dosage Forms and Strengths
3 DOSAGE FORMS AND STRENGTHS
For injection: 500 mg of deferoxamine mesylate (corresponding to 426.82 mg of deferoxamine as free base) as a white to off-white lyophilized powder in single-dose fliptop vial for reconstitution.
For injection: 2 g of deferoxamine mesylate (corresponding to 1707.28 mg of deferoxamine as free base) as a white to off-white lyophilized powder in single-dose fliptop vial for reconstitution.
Contraindications
4 CONTRAINDICATIONS
Deferoxamine Mesylate for injection is contraindicated in patients with:
- •
- A history of a hypersensitivity reaction to deferoxamine or any of its inactive ingredients [see Description (11)]. Reactions have included anaphylaxis [see Warnings and Precautions (5.1)].
- •
- Severe renal disease or anuria since the drug and the iron chelate are excreted primarily by the kidney [see Warnings and Precautions (5.3)].
Warnings and Precautions
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis, have occurred in deferoxamine mesylate-treated patients. Reactions have included flushing of the skin, urticaria, hypotension, and shock. These reactions typically occur when deferoxamine mesylate was administered by rapid intravenous injection. Therefore, administer deferoxamine mesylate intramuscularly or by slow subcutaneous or intravenous infusion.
5.2 Auditory and Ocular Toxicity
Ocular and auditory toxicities have been reported in deferoxamine mesylate-treated patients. The ocular toxicities observed have included blurring of vision; cataracts after prolonged administration in chronic iron overload; decreased visual acuity, including visual loss, visual defects, scotoma; impaired peripheral, color, and night vision; optic neuritis, cataracts, corneal opacities, and retinal pigmentary abnormalities. The auditory toxicities reported have been tinnitus and hearing loss, including high frequency sensorineural hearing loss. Risk factors for both ocular and auditory disturbances include prolonged treatment duration, higher doses, or low ferritin levels. In most cases, both ocular and auditory disturbances were reversible upon immediate cessation of treatment [see Adverse Reactions (6)].
Visual acuity tests, slit-lamp examinations, funduscopy and audiometry are recommended periodically in patients treated for prolonged periods of time. Toxicity is more likely to be reversed if symptoms or test abnormalities are detected early.
5.3 Renal Toxicity
Renal toxicity, including increases in serum creatinine (possibly dose-related), acute renal failure and renal tubular disorders has occurred in deferoxamine mesylate-treated patients. Deferoxamine mesylate is contraindicated in patients with severe renal disease [see Contraindications (4)]. Monitor serum creatinine to assess for changes in renal function.
5.4 Respiratory Toxicity
Acute respiratory distress syndrome has occurred in deferoxamine mesylate-treated patients following treatment with excessively high intravenous doses of deferoxamine mesylate in patients with acute iron intoxication or thalassemia. The recommended daily doses should therefore not be exceeded.
5.5 Growth Suppression
High doses of deferoxamine mesylate and concomitant low ferritin levels have also been associated with growth suppression in pediatric patients. After reduction of deferoxamine mesylate dose, growth velocity may partially resume to pre‑treatment rates. Monitor growth (weight and height) in pediatric patients treated with deferoxamine mesylate every 3 months.
5.6 Serious Infections
Yersinia Infections
Deferoxamine mesylate may increase the risk of Yersinia enterocolitica and Yersinia pseudotuberculosis infections. Avoid starting Deferoxamine mesylate treatment in patients with active Yersinia infections. Should Yersinia infection develop, interrupt deferoxamine mesylate treatment until the infection is resolved.
Mucormycosis
Cases of mucormycosis, some with a fatal outcome, have occurred in deferoxamine mesylate-treated patients. Signs or symptoms are specific to the site of infection. If mucormycosis is suspected, discontinue deferoxamine mesylate, conduct mycological testing, and treat immediately.
5.7 Cardiac Dysfunction with Concomitant Use of Vitamin C
Cardiac dysfunction has occurred in deferoxamine mesylate-treated patients with severe chronic iron overload following concomitant treatment with high doses of vitamin C (more than 500 mg daily in adults). The cardiac dysfunction was reversible when vitamin C was discontinued. The following precautions should be taken when vitamin C and deferoxamine mesylate are to be used concomitantly:
- •
- Vitamin C supplements should not be given to patients with cardiac failure.
- •
- Start supplemental vitamin C only after an initial month of regular treatment with deferoxamine mesylate.
- •
- Give vitamin C only if the patient is receiving deferoxamine mesylate regularly, ideally soon after setting up the infusion pump.
- •
- Do not exceed a daily vitamin C dose of 200 mg in adults, given in divided doses. In general, 50 mg daily suffices for pediatric patients under 10 years old and 100 mg for older pediatric patients.
- •
- Clinical monitoring of cardiac function is advisable during such combined therapy.
5.8 Risks of Deferoxamine Mesylate Treatment in Patients with Aluminum Overload
Deferoxamine mesylate may cause neurological dysfunction (including seizures) in patients with aluminum‑related encephalopathy and receiving dialysis, possibly due to an acute increase in circulating aluminum [see Adverse Reactions (6)].
Deferoxamine mesylate may precipitate the onset of dialysis dementia.
Treatment with deferoxamine mesylate in the presence of aluminum overload may result in decreased serum calcium and aggravation of hyperparathyroidism.
5.9 Effects on Ability to Drive and Use Machines
Deferoxamine mesylate may cause dizziness, which may impair the ability to drive a car or operate machinery. Patients should not drive or operate machinery until they know how deferoxamine mesylate will affect their ability to engage in these activities.
5.10 Embryo-Fetal Toxicity
Based on findings in animals, deferoxamine mesylate can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of deferoxamine to pregnant mice and rabbits during the period of organogenesis caused adverse developmental outcomes including decreased fetal body weights and malformations at maternal doses less than those in patients at maximum recommended human dose (MRHD). Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment with deferoxamine mesylate and for one month after the last dose [see Use in Specific Populations (8.1, 8.3), Nonclinical Toxicology (13.1)].
Adverse Reactions
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- •
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- •
- Auditory and Ocular Toxicity [see Warnings and Precautions (5.2)]
- •
- Renal Toxicity [see Warnings and Precautions (5.3)]
- •
- Respiratory Toxicity [see Warnings and Precautions (5.4)]
- •
- Growth Suppression [see Warnings and Precautions (5.5)]
- •
- Serious Infections [see Warnings and Precautions (5.6)]
- •
- Cardiac Dysfunction with Concomitant Use of Vitamin C [see Warnings and Precautions (5.7)]
- •
- Risks of Deferoxamine mesylate Treatment in Patients with Aluminum Overload [see Warnings and Precautions (5.8)]
- •
- Effects on Ability to Drive and Use Machines [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
The following adverse reactions associated with the use of Deferoxamine mesylate were identified in clinical studies or postmarketing reports. Because some of these reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
At the Injection Site: Localized irritation, pain, burning, swelling, induration, infiltration, pruritus, erythema, wheal formation, eschar, crust, vesicles, local edema. Injection site reactions may be associated with systemic allergic reactions (see Body as a Whole, below)
Hypersensitivity Reactions and Systemic Allergic Reactions: Generalized rash, urticaria, anaphylactic reaction with or without shock, angioedema
Body as a Whole: Local injection site reactions may be accompanied by systemic reactions like arthralgia, fever, headache, myalgia, nausea, vomiting, abdominal pain, or asthma
Infections: Yersinia, mucormycosis
Cardiovascular: Tachycardia, hypotension, shock
Digestive: Abdominal discomfort, diarrhea, nausea, vomiting
Hematologic: Blood dyscrasia (thrombocytopenia, leukopenia)
Hepatic: Increased transaminases, hepatic dysfunction
Musculoskeletal: Muscle spasms. Growth retardation and bone changes (e.g., metaphyseal dysplasia)
Nervous System: Neurological disturbances, including dizziness, peripheral sensory, motor, or mixed neuropathy, paresthesias, seizures; exacerbation or precipitation of aluminum-related dialysis encephalopathy
Special Senses: High-frequency sensorineural hearing loss, tinnitus, visual disturbances including acuity, blurred vision, loss of vision, dyschromatopsia, night blindness, visual field defects, scotoma, retinopathy (pigmentary degeneration), optic neuritis, and cataracts
Respiratory: Acute respiratory distress syndrome (with dyspnea, cyanosis, and/or interstitial infiltrates)
Skin: Generalized rash
Urogenital: Dysuria, acute renal failure, increased serum creatinine and renal tubular disorders
Drug Interactions
Use in Specific Populations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on Deferoxamine mesylate use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriages or adverse maternal or fetal outcomes.
In animal reproduction studies subcutaneous administration of deferoxamine to pregnant animals (mice or rabbits) during organogenesis at doses approximately ≥0.2- (mice) and ≥0.7 (rabbits) times the maximum recommended human dose resulted in maternal toxicity and adverse developmental outcomes (see Data). Advise pregnant women of the potential risk to a fetus. Consider the benefits and risks of Deferoxamine mesylate for the mother and possible risks to the fetus when prescribing Deferoxamine mesylate to a pregnant woman.
The estimated background risk of major birth defects and miscarriage for the indicated population(s) is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Animal Data
In an embryo-fetal developmental study in mice, pregnant animals administered subcutaneous doses of deferoxamine at 180, and 540 mg/kg/day from gestation day 7 to gestation day 12 resulted in a dose dependent delay and irregularities of fetal skeletal maturation at doses ≥0.2 times the MRHD. At the highest dose of 540 mg/kg, in 1/23 fetuses had a unilateral lesion to the eye lens (approximately 0.5 times the MRHD).
In the embryo-fetal developmental studies in rabbits, pregnant animals administered subcutaneous doses of deferoxamine either 200 mg/kg or 200, 300, and 540 mg/kg from gestation day 6 to gestation day 14 resulted in maternal toxicity and embryo-fetal developmental effects at 0.7 times the MRHD. Maternal toxicity included reduced fetal body weights and embryo-fetal effects included malformations of spina bifida, and increased incidence of abnormally ossified ribs and vertebrae.
No maternal toxicity or embryo-fetal effects were observed in rats at deferoxamine doses tested (up to 0.9 times the MRHD).
8.2 Lactation
There are no data on the presence of deferoxamine or its metabolite in either human or animal milk, the effects on the breastfed child, or the effects on milk production. It is not known whether deferoxamine is excreted in human milk. Because of the potential for serious adverse reactions in the breastfed child, advise patients not to breastfeed during treatment with Deferoxamine mesylate, and for one week after the last dose.
8.3 Females and Males of Reproductive Potential
Based on animal data, Deferoxamine mesylate can cause malformations at doses less than the human dose [see Use in Specific Populations (8.1)].
Contraception
Females
Deferoxamine mesylate can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise female patients of reproductive potential to use effective contraception during treatment with Deferoxamine mesylate and for one month after the last dose.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients 3 years of age and older have been established for the treatment of acute iron intoxication and for the treatment of transfusional iron overload in patients with chronic anemia. Safety and effectiveness in pediatric patients under the age of 3 years have not been established.
Iron mobilization with Deferoxamine mesylate is relatively poor in patients under the age of 3 years with relatively little iron overload. Deferoxamine mesylate is not recommended for use. The drug should ordinarily not be given to these patients unless significant iron mobilization (e.g., 1 mg or more of iron per day) can be demonstrated.
High doses of Deferoxamine mesylate and concomitant low ferritin levels have been associated with growth suppression in pediatric patients. Monitor weight and height in pediatric patients receiving Deferoxamine mesylate every 3 months [see Warnings and Precautions (5.5), Adverse Reactions (6.1)].
8.5 Geriatric Use
Clinical Studies of deferoxamine mesylate did not include sufficient numbers of subjects aged 65 years and over to determine whether they respond differently from the younger subjects. Postmarketing reports suggest a possible trend for an increased risk of eye disorders in the geriatric population, specifically the occurrence of color blindness, maculopathy, and scotoma. However, it is unclear if these eye disorders were dose related. Although the number of reports was very small, certain elderly patients may be predisposed to eye disorders when taking deferoxamine mesylate. Postmarketing reports also suggest that there may be an increased risk of deafness and hearing loss in the geriatric population [see Adverse Reactions (6)]. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Renal Impairment
Deferoxamine mesylate is contraindicated in patients with severe renal disease [see Contraindications (4)].
For patients with renal impairment, dose selection should usually start at the low end of the dosing range.
Deferoxamine can cause increases in serum creatinine (possibly dose-related), acute renal failure and renal tubular disorders [see Warnings and Precautions (5.3)]. Monitor patients for changes in renal function.
Overdosage
10 OVERDOSAGE
Acute Toxicity
Intravenous LD50s (mg/kg): mice, 287; rats, 329.
Inadvertent administration of an overdose or inadvertent intravenous bolus administration/rapid intravenous infusion may be associated with hypotension, tachycardia and gastrointestinal disturbances; acute but transient loss of vision, aphasia, agitation, headache, nausea, pallor, CNS depression, including coma, bradycardia and acute renal failure have been reported.
Acute respiratory distress syndrome has been reported following treatment with excessively high intravenous doses of Deferoxamine Mesylate for injection in patients with acute iron intoxication and in patients with thalassemia.
There is no specific antidote for Deferoxamine mesylate overdose. In case of overdose, discontinue Deferoxamine mesylate and provide symptomatic supportive care.
Deferoxamine mesylate is readily dialyzable.
Description
11 DESCRIPTION
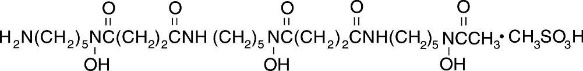
Deferoxamine Mesylate for Injection, USP, is an iron-chelating agent, available in vials for injection via intramuscular, subcutaneous, and intravenous administration. Deferoxamine mesylate is supplied as vials containing 500 mg of deferoxamine mesylate USP (corresponding to 426.82 mg of deferoxamine as free base) and 2 g of deferoxamine mesylate USP (corresponding to 1707.28 mg of deferoxamine as free base) in sterile, lyophilized form. Deferoxamine mesylate is N-[5-[3-[(5-aminopentyl)hydroxycarbamoyl]propionamido]pentyl]-3-[[5-(N-hydroxyacetamido)pentyl]carbamoyl]propionohydroxamic acid monomethanesulfonate (salt), and its structural formula is:
Deferoxamine mesylate USP is a white to off-white powder. It is freely soluble in water and slightly soluble in methanol. Its molecular weight is 656.79 g/mol.
Clinical Pharmacology
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Deferoxamine mesylate chelates iron by forming a stable complex that prevents the iron from entering into further chemical reactions. It readily chelates iron from ferritin and hemosiderin but not readily from transferrin; it does not combine with the iron from cytochromes and hemoglobin.
Deferoxamine mesylate does not cause any demonstrable increase in the excretion of electrolytes or trace metals. Theoretically, 100 parts by weight of deferoxamine mesylate is capable of binding approximately 8.5 parts by weight of ferric iron.
12.3 Pharmacokinetics
Deferoxamine mesylate is metabolized principally by plasma enzymes, but the pathways have not yet been defined. The chelate is readily soluble in water and passes easily through the kidney, giving the urine a characteristic reddish color. Some is also excreted in the feces via the bile.
Nonclinical Toxicology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies in animals have not been performed with deferoxamine mesylate. Cytotoxicity may occur, since deferoxamine mesylate has been shown to inhibit DNA synthesis in vitro.
Deferoxamine mesylate was not mutagenic when tested in an in vitro bacterial reverse mutation (Ames) and was not genotoxic in an in vivo micronucleus assay in rats.
Animal studies to assess fertility effects have not been conducted.
How Supplied/Storage and Handling
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Unit of Sale | Concentration |
NDC 0409-2336-10 Carton of 4 Single-dose Fliptop vials | 500 mg |
NDC 0409-2337-25 Carton of 4 Single-dose Fliptop vials | 2 g |
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F) excursions permitted between 15°C and 30°C (59°F and 86°F).
Discard unused portion.
Patient Counseling Information
17 PATIENT COUNSELING INFORMATION
Caution patients about the potential allergic reactions associated with rapid intravenous administration of Deferoxamine Mesylate for injection and the need for monitoring allergic reactions during treatment [see Warnings and Precautions (5.1)].
Caution patients about the potential auditory and ocular toxicities due to prolonged use of Deferoxamine Mesylate for injection, conduct auditory testing and ophthalmic testing at regular intervals. Advise patients to contact their healthcare provider if they develop visual or auditory changes during treatment [see Warnings and Precautions (5.2)].
Caution patients about the potential for kidney toxicity when taking Deferoxamine Mesylate for injection and the need for kidney function test to monitor for increase in serum creatinine [see Warnings and Precautions (5.3)].
Inform patients that if they have difficulty in breathing during treatment, they should inform the health care provider as this is a symptom of acute respiratory distress syndrome which can occur with excessively high intravenous doses of Deferoxamine Mesylate for injection [see Warnings and Precautions (5.4)].
Caution pediatric patients and their caregivers that child treated with Deferoxamine Mesylate for injection could have slower than normal growth and the need to monitor for body weight and height every 3 months [see Warnings and Precautions (5.5)].
Caution patients about the increased risk of bacterial infections (Yersinia enterocolitica and Yersinia pseudotuberculosis) with Deferoxamine Mesylate for injection treatment and the need for treatment discontinuation until the infection is resolved [see Warnings and Precautions (5.6)].
Caution patients about the potential risk of fungal infections (Mucormycosis) when receiving Deferoxamine Mesylate for injection treatment and the need for treatment discontinuation, mycological tests and required treatment for treating the infection [see Warnings and Precautions (5.6)].
Caution patients about the potential impairment of cardiac function when taking Deferoxamine Mesylate for injection concomitantly with high doses of Vitamin C (more than 500 mg daily in adults). Inform adult patients not to exceed a daily Vitamin C dose of 200 mg given in divided doses. Inform pediatric patients under 10 years of age and older pediatric patients or their care takers not to exceed a daily Vitamin C of 50 mg and 100 mg, respectively [see Dosage and Administration (2.4) and Warnings and Precautions (5.7)].
Inform patients with cardiac failure not to take Vitamin C supplements when on treatment with Deferoxamine Mesylate for injection [see Dosage and Administration (2.4) and Warnings and Precautions (5.7)].
Caution patients with aluminum-related encephalopathy and receiving dialysis about potential neurological dysfunction [see Warnings and Precautions (5.8)].
Cautions patients that treatment with Deferoxamine Mesylate for injection in the presence of aluminum overload may result in decreased serum calcium and aggravation of hyperparathyroidism [see Warnings and Precautions (5.8)].
Inform patients that they should refrain from driving or operating potentially hazardous machines if they experience dizziness or other nervous system disturbances, or impairment of vision or hearing [see Warnings and Precautions (5.9)].
Advise patients to inform the healthcare provider if they have received prochlorperazine prior to Deferoxamine Mesylate for injection treatment as this may lead to temporary impairment of consciousness [see Drug Interactions (7.1)].
Inform patients that if they are going for any imaging tests while receiving Gallium-67 and Deferoxamine mesylate concomitantly it can result in reports with distorted images [see Drug Interactions (7.2)].
Inform patients that their urine may occasionally show a reddish discoloration.
Embryo-fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.10), Use in Specific Populations (8.1, 8.3)].
Advise females of reproductive potential to use effective contraceptive during treatment with Deferoxamine Mesylate for injection and for one month after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise patients to avoid breastfeeding while taking Deferoxamine Mesylate for injection and for one week after the final dose [see Use in Specific Populations (8.2)].
Distributed by Hospira, Inc., Lake Forest, IL 60045 USA 
LAB-1006-5.0
17 PATIENT COUNSELING INFORMATION
17 PATIENT COUNSELING INFORMATION
Caution patients about the potential allergic reactions associated with rapid intravenous administration of Deferoxamine Mesylate for injection and the need for monitoring allergic reactions during treatment [see Warnings and Precautions (5.1)].
Caution patients about the potential auditory and ocular toxicities due to prolonged use of Deferoxamine Mesylate for injection, conduct auditory testing and ophthalmic testing at regular intervals. Advise patients to contact their healthcare provider if they develop visual or auditory changes during treatment [see Warnings and Precautions (5.2)].
Caution patients about the potential for kidney toxicity when taking Deferoxamine Mesylate for injection and the need for kidney function test to monitor for increase in serum creatinine [see Warnings and Precautions (5.3)].
Inform patients that if they have difficulty in breathing during treatment, they should inform the health care provider as this is a symptom of acute respiratory distress syndrome which can occur with excessively high intravenous doses of Deferoxamine Mesylate for injection [see Warnings and Precautions (5.4)].
Caution pediatric patients and their caregivers that child treated with Deferoxamine Mesylate for injection could have slower than normal growth and the need to monitor for body weight and height every 3 months [see Warnings and Precautions (5.5)].
Caution patients about the increased risk of bacterial infections (Yersinia enterocolitica and Yersinia pseudotuberculosis) with Deferoxamine Mesylate for injection treatment and the need for treatment discontinuation until the infection is resolved [see Warnings and Precautions (5.6)].
Caution patients about the potential risk of fungal infections (Mucormycosis) when receiving Deferoxamine Mesylate for injection treatment and the need for treatment discontinuation, mycological tests and required treatment for treating the infection [see Warnings and Precautions (5.6)].
Caution patients about the potential impairment of cardiac function when taking Deferoxamine Mesylate for injection concomitantly with high doses of Vitamin C (more than 500 mg daily in adults). Inform adult patients not to exceed a daily Vitamin C dose of 200 mg given in divided doses. Inform pediatric patients under 10 years of age and older pediatric patients or their care takers not to exceed a daily Vitamin C of 50 mg and 100 mg, respectively [see Dosage and Administration (2.4) and Warnings and Precautions (5.7)].
Inform patients with cardiac failure not to take Vitamin C supplements when on treatment with Deferoxamine Mesylate for injection [see Dosage and Administration (2.4) and Warnings and Precautions (5.7)].
Caution patients with aluminum-related encephalopathy and receiving dialysis about potential neurological dysfunction [see Warnings and Precautions (5.8)].
Cautions patients that treatment with Deferoxamine Mesylate for injection in the presence of aluminum overload may result in decreased serum calcium and aggravation of hyperparathyroidism [see Warnings and Precautions (5.8)].
Inform patients that they should refrain from driving or operating potentially hazardous machines if they experience dizziness or other nervous system disturbances, or impairment of vision or hearing [see Warnings and Precautions (5.9)].
Advise patients to inform the healthcare provider if they have received prochlorperazine prior to Deferoxamine Mesylate for injection treatment as this may lead to temporary impairment of consciousness [see Drug Interactions (7.1)].
Inform patients that if they are going for any imaging tests while receiving Gallium-67 and Deferoxamine mesylate concomitantly it can result in reports with distorted images [see Drug Interactions (7.2)].
Inform patients that their urine may occasionally show a reddish discoloration.
Embryo-fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.10), Use in Specific Populations (8.1, 8.3)].
Advise females of reproductive potential to use effective contraceptive during treatment with Deferoxamine Mesylate for injection and for one month after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise patients to avoid breastfeeding while taking Deferoxamine Mesylate for injection and for one week after the final dose [see Use in Specific Populations (8.2)].
Distributed by Hospira, Inc., Lake Forest, IL 60045 USA
LAB-1006-5.0
Resources
Didn’t find what you were looking for?
Contact us.
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine.
*Speak with a Pfizer Medical Information Professional regarding your medical inquiry. Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for Pfizer prescription products.
Report Adverse Event
Pfizer Safety
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this product, click the link below to submit your information: Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer product, please call Pfizer Medical Information at (800) 438-1985.
FDA Medwatch
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800)-332-1088.